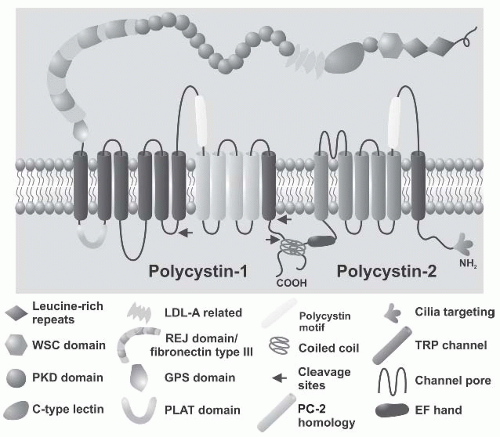
INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD; MIM 173990) is the most common hereditary renal disease occurring in 1:400 to 1:1000 individuals. It accounts for over 90% of all hereditary renal cystic diseases (
Table 16.1). ADPKD is characterized by the presence of bilateral renal cysts that gradually grow and expand over time, resulting in significantly increased total kidney volume, progressive renal injury, and ultimately, end-stage renal disease (ESRD), usually in the sixth decade of life.
Other Mendelian diseases with varying degrees of fibrocystic involvement of the kidney are relatively rare. Autosomal recessive polycystic kidney disease (ARPKD; MIM 606702) occurs in 1:25,000; autosomal dominant tuberous sclerosis complex (TSC; MIM 191100 and 613254) occurs in 1:10,000; autosomal dominant medullary cystic disease or UMOD (MIM 174000) occurs in 1:35,000; and autosomal recessive familial juvenile nephronophthisis (NPHP; MIM 256100 and 602088) occurs in 1:40,000. In addition to the Mendelian diseases described previously, a variety of hereditary syndromes, such as Bardet-Biedl syndrome (BBS; MIM 209900) and Meckel Gruber syndrome (MKS; MIM 249000), result in a constellation of clinical manifestations that include renal cysts and are collectively termed ciliopathies (
Table 16.1). In terms of clinical disease burden, however, all of the hereditary ciliopathies combined account for less than 1% of all renal cystic diseases.
This chapter will focus primarily on the single most common renal cystic disease, ADPKD. Current understanding of the epidemiology, clinical characteristics, and the pathology of ADPKD will be elucidated, and the appropriate approaches to the diagnosis and management of ADPKD will be provided. An overview of the genetics and the molecular pathways involving the polycystins is provided. Finally, a review of molecularly targeted therapeutic interventional trials will be presented to establish the potential future management for ADPKD individuals.
EPIDEMIOLOGY
Worldwide, ADPKD occurs between 1:400 and 1:1000 live births in all ethnicities when including ascertainment by autopsy.
1,
2,
3 Epidemiologic studies suggest that fewer than half of those with the disease are diagnosed during their life, with a majority of diagnoses occurring on autopsy after death from other causes.
1 Although not specifically evaluated in those studies, it is plausible that these individuals had a milder manifestation of disease. ADPKD has been estimated to be present in up to 600,000 individuals in the United States of America, and 12.5 million people worldwide (www .pkdcure.org). The disease accounts for about 6% of all patients on hemodialysis.
4 The incidence of ADPKD varies by genotype.
PKD1 occurs in approximately 1:700 live births and
PKD2 occurs in 1:15,000 live births. Reports from Japan, Denmark, the United States, Europe, India, Saudi Arabia, and Turkey show similar incidence rates, with no racial or ethnic predilection.
5,
6,
7,
8,
9 Patient and renal outcomes differ by PKD genotype. ADPKD resulting from mutations in
PKD1 result in earlier mean age of onset of hypertension (29 versus 41 years of age), ESRD (55 versus 74 years of age), and death (68 versus 79 years of age) when compared to ADPKD due to
PKD2 mutations.
10,
11,
12Current worldwide yearly incidence rates for ESRD due to ADPKD are 7.5 and 6.1 per million population for men and women, respectively. Gender differences with regard to disease progression and severity have been reported, with women having a more favorable renal outcome than men.
10,
13 However, when gender differences and genotype are considered together, little or no differences in the age of onset of renal failure are found in
PKD1 individuals. Importantly, significant differences in survival are found between men and women with
PKD2 mutations (67.3 versus 71.0 years). In addition, previous age-adjusted sex ratios of the incidence of ESRD, which were greater in men than in women (1.4 to 1.6), are now beginning to reach parity in various countries, including Denmark and the United States.
African American ADPKD patients have been reported to have a more aggressive renal course than their non-African American counterparts.
14,
15,
16 However, these reports are limited due to the small number of patients and the presence of other diseases affecting renal function, including the sickle cell disease or trait. Importantly, the relative incidence of ESRD due to ADPKD per million population in African Americans is lower than their non-African American counterparts.
4,
17,
18 Whether this relates to earlier mortality in African Americans or improved renal survival has not yet been determined.
During the past 3 decades, the average age of onset of ESRD, the incidence and prevalence rates of ADPKD in ESRD, and the survival rates of ADPKD in ESRD have increased. Incidence rates of ADPKD patients entering ESRD have increased significantly from 1990 to 2010 in the United States (35%), Japan (30%), and Denmark (33%).
5,
17,
19 The average age of onset of ESRD has increased in the United States (4.5 years), Denmark (5.1 years), and Japan (4.6 years). Importantly, the use and the number of antihypertensive agents, specifically inhibitors of the renin-angiotensin-aldosterone system (RAAS) are associated with decreased patient mortality and an increased age of onset of ESRD.
20,
21 Taken together, these data suggest that patients are now more often surviving to start renal replacement therapy, and improved patient care has extended both renal and patient survival with a positive impact on patient mortality for those receiving renal replacement therapy.
DIAGNOSIS OF AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE
Although symptoms in ADPKD individuals can present at a young age, a diagnosis of ADPKD is confirmed either with radiographic imaging or genetic testing. Before undertaking a diagnostic evaluation, counseling should be done to educate the family about the risk for inheritance. The mode of presentation for diagnostic screening has not changed over the last half century, with approximately 40% of individuals with a positive family history presenting asymptomatically.
13,
22 The remaining patients present with clinical complications such as new onset hypertension, gross hematuria, acute flank pain, or fevers. The average age of presentation for diagnosis has also remained unchanged for the last 4 decades. With a number of potential beneficial therapies becoming available and evidence for improved mortality due to standard medical care, an earlier presentation for screening of at risk individuals will most likely occur.
The risk for discrimination in terms of insurability and employment has been reduced, but not eliminated, by the passage into law of the Genetic Information Nondiscrimination Act (GINA).
23,
24 GINA prohibits insurers from canceling, denying, refusing to renew, or changing the terms or premiums of coverage based on genetic information. It also prohibits employers from making hiring, firing, promotion, and other employment-related decisions based on genetic factors. Genetic information is defined as information about an individual’s genetic tests, the genetic tests of family members, or occurrence of a disease in family members of the individual. GINA, however, applies only to individuals who are asymptomatic, does not prohibit underwriting based on information about current health status, and does not apply to life insurance, disability insurance, or long-term care insurance.
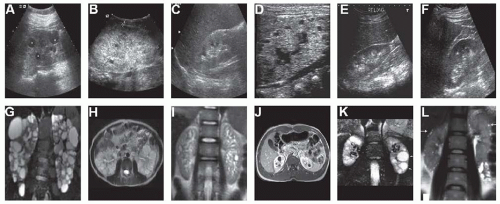
Ultrasound imaging is the initial imaging modality of choice for a diagnosis of ADPKD (
Fig. 16.1). Ultrasound is cost-efficient as compared to magnetic resonance imaging (MRI) or computed tomography (CT) and is not associated with the radiation exposure that occurs with CT imaging. The vast majority of affected ADPKD patients can be diagnosed by ultrasound imaging alone. Since the initial ultrasound criteria for a diagnosis of ADPKD were developed in 1994,
25 the imaging resolution for detection of small cysts with ultrasound, CT, and MRI have vastly improved.
26,
27 The limit of detection of cysts using CT and MRI is now as low as 1 mm in diameter as compared to 0.5 to 1 cm with ultrasound.
28 Importantly, and relevant to the value of diagnostic imaging in ADPKD, simple renal cysts
>1 cm in diameter remain relatively rare in childhood, occurring in <0.1% in the general population.
MR- and CT-based imaging studies of healthy young adults without a family history of ADPKD show that simple renal cysts of diameters as small as 1 mm are relatively common, even in young adults, occurring in 11 out of 35 or 28% in 18- to 29-year-olds and 97 out of 190 or 51% in 30- to 44-year-olds.
26 Therefore, if Ravine criteria using cyst number alone is used with CT or MRI in individuals between the ages of 18 and 45 years of age, more than one third would erroneously qualify for a diagnosis of ADPKD. However, if only those with cysts
>1 cm in diameter are considered, the size of the cyst commonly seen in ADPKD individuals and detectable by ultrasonography, the number of incorrectly diagnosed individuals would remain <1%. Therefore, in at-risk individuals, sensitivity and specificity for a correct diagnosis of ADPKD using ultrasound remains intact and a single renal cyst in an at-risk child from a family with AD-PKD is sufficient to make a diagnosis (
Table 16.2).
29An age-based renal cyst number is required for a diagnosis of ADPKD, given that simple renal cysts are present with increasing frequency as age increases in the general population. Ultrasound still carries high sensitivity and specificity for the majority of
PKD1 individuals over the age of 30 years.
30,
31 At age 30, a negative ultrasound indicates a less than 5% likelihood of having ADPKD in individuals from
PKD1 families. Negative ultrasound imaging is also informative at earlier ages in at-risk individuals from
PKD1 families, with a negative ultrasound in an at-risk 20-year-old conferring a less than 10% chance of carrying the disease. Additional experience with screening
PKD2 individuals provides a more accurate estimate of the relatively high falsenegative rates when screening at-risk individuals under the age of 40.
32 Although the specificity and positive predictive value of sonographic criteria is very high in
PKD1 individuals, their sensitivity and negative predictive value are low when applied to
PKD2 in the 15 to 29 age group (69.5 and 78%, respectively). This is particularly a problem when evaluating potential young related kidney transplant donors, where the exclusion of ADPKD is important.
Based on this experience, Pei and colleagues
33 are now able to provide a unified age-based criteria for a diagnosis of ADPKD using ultrasound imaging for both
PKD1 and
PKD2 individuals (
Table 16.2). This modification from the original Ravine criteria increases the age from 30 to 40 years for screening purposes. The presence of at least three (unilateral or bilateral) renal cysts and two cysts in each kidney are sufficient for a diagnosis of both at-risk individuals and those without a family history of ADPKD aged 15 to 39 years and 40 to 59 years, respectively. The requirement of three or more cysts (unilateral or bilateral) has a positive predictive value of 100% in the younger age group and minimizes false-positive diagnoses, because 2.1% and 0.7% of unaffected healthy individuals younger than 30 years have one and two renal cysts, respectively. In those 30 to 39 years old, both the original (two cysts in each kidney) and the revised (three cysts, unilateral or bilateral) criteria have a positive predictive value of 100%. Finally, for at-risk individuals aged greater than 60 years, four or more cysts in each kidney are required. Even with these criteria in place, there are still exceptions where ultrasound screening is not sufficient, usually when either a family history for ADPKD is absent and the clinical presentation is atypical, or when clinical suspicion for a positive diagnosis is high and evidence for renal cystic disease by ultrasound is lacking.
Although a minimum number of renal cysts are required for a diagnosis of ADPKD, other renal cystic diseases may also meet the requisite cyst number criteria to qualify for a diagnosis of ADPKD (
Fig. 16.1). Additionally, given that approximately 15% of ADPKD individuals develop polycystic kidney disease (PKD) spontaneously and have unaffected biologic parents, other features of the clinical presentation are important to consider beyond simply the number of renal cysts identified. For example, the distribution and size of cysts can be informative. Cysts occurring predominantly in the medullary space with relatively small size are found in medullary cystic disease and familial juvenile nephronophthisis in the setting of normal or small kidney size and can be used to distinguish these from ADPKD. Angiomyolipomas are typically present in the kidneys of patients with tuberous sclerosis complex. Glomerular cystic disease is associated with relatively small widely distributed discrete cysts found predominantly in the cortex in the setting of normal kidney size.
In contrast to many other hereditary renal cystic diseases, ADPKD is characterized by significant renal enlargement in the setting of normal kidney function. Although renal enlargement is a feature of ARPKD, particularly in those diagnosed in utero or at birth, significant renal insufficiency usually accompanies this feature. The cysts in ARPKD tend to be diffuse, small, and relatively homogeneous and are more commonly described as fusiform and ectactic dilatations rather than discrete macrocysts. Significant renal enlargement is not present in other hereditary kidney diseases including von Hippel Lindau disease, nephronophthisis,
and medullary cystic disease. Renal enlargement with significant renal cystic burden in the setting of normal kidney function, often with early onset, can be seen in specific individuals who have contiguous gene deletion mutations involving both the
PKD1 and
TSC2 genes.
34 This contiguous gene syndrome usually requires further genetic testing for accurate diagnosis. In addition to the diagnostic value of kidney enlargement in the setting of normal kidney function in ADPKD, the presence of radiographically visible liver cystic disease, when present, is also a unique and diagnostically useful feature of ADPKD. No other hereditary renal cystic disease is accompanied by polycystic liver disease with the exception of some instances of familial juvenile nephronophthisis. Importantly, familial instances of liver cystic disease indistinguishable from that seen in ADPKD but lacking kidney cysts is a genetically distinct disorder.
35Genetic testing may be required to confirm or exclude a diagnosis of ADPKD. Genetic testing is reserved for patients with renal cystic disease without a family history and with an uncertain presentation of ADPKD, or those with a negative ultrasound who are at risk for ADPKD but who need a confirmatory diagnosis for the purposes of living related kidney donation, family planning, or for occupational safety. Direct sequencing of the
PKD1 and
PKD2 genes is the most reliable approach to a genetic diagnosis.
36,
37,
38 A curated database of
PKD1 and
PKD2 mutations is available at
http://pkdb.mayo .edu/. Mutation detection is successful in up to 85% of cases in research laboratories. Destructive mutations (i.e., those predicted to result in truncated proteins due to premature termination codons, aberrant splicing, or insertion-deletions resulting in frame shifting) are readily identifiable as pathogenic. The same is not true for mutations due to nonsynonymous amino acid substitutions, which may account for up to 30% of mutations in
PKD1 and a significantly lower percentage of
PKD2. The pathogenicity of missense sequence variations often need to be confirmed with segregation studies in other affected family members before a diagnosis can be confirmed. A genetic diagnosis is further complicated by the lack of commonly recurring mutations that have been identified in other diseases such as cystic fibrosis. As a result, most families have private mutations requiring the relatively expensive whole gene sequencing approach for detection that nonetheless yields a mutation detection rate (highest detection rate, 85%) lower than the rate of cyst detection by age-appropriate ultrasound (lowest detection rate, 99%). As a result, this approach is reserved in the clinical setting for a limited group of patients meeting the previous criteria.
PATHOLOGY OF POLYCYSTIC KIDNEY DISEASE
The kidneys of patients with polycystic disease gradually enlarge and attain an enormous size due to the growth of hundreds of cysts. Kidneys measuring 40 ×25 ×20 cm and weighing 7 to 8 kg have been reported. Usually, these greatly enlarged kidneys are seen in patients with ESRD undergoing
nephrectomy or at autopsy. These end-stage kidneys contain hundreds of fluid-filled cysts of widely differing sizes. The cyst walls can be thin and transparent, but calcification of cyst walls is also common.
39 The renal capsule may be thickened around infected cysts, and the kidney may be attached to adjacent abdominal organs such as the spleen and the adrenal glands by fibrous tissue.
Cut sections demonstrate cysts throughout the renal parenchyma. Islands of normal-appearing renal parenchyma can usually be found only in kidneys from young, nonazotemic patients. The cysts vary in size from 1 mm to 10 cm or more in diameter. Cysts in ADPKD arise from all segments of the nephron, and some cysts (˜11%) retain the morphologic characteristics of proximal or distal tubules or collecting ducts. However, most (84%) are lined by a single layer of poorly differentiated columnar or cuboidal epithelium.
40,
41 Approximately 5% of cysts are lined by a markedly hyperplastic epithelium, forming polyps and microadenomas.
40,
42 This hyperproliferative epithelium typically has no signs of dysplasia or premalignant features. The cysts are surrounded by a fibrous stroma, which may contain bundles of smooth musclelike cells, likely transformed myofibroblasts. Inflammatory interstitial infiltrates are seen, and in advanced cases, the renal interstitium is replaced by fibrosis. Marked arteriosclerosis and arteriolosclerosis are found in nephrectomy specimens, evidence that ischemic injury and damage from hypertension contribute to tubular atrophy and glomerulosclerosis.
43Microdissection studies of human kidneys suggest that only 1% to 2% of nephrons are cystic.
40 These studies also have shown that cysts begin as focal dilatations of tubular segments.
44 When these dilatations exceed approximately 2 mm in diameter, they typically disconnect from the parent tubule; at least 73% of the cysts have no tubular openings when evaluated by scanning electron microscopy.
40 The cysts lining epithelial cells are joined together by junctional complexes like those seen in normal proximal tubules or by tight junctions typical of the distal renal epithelium.
41 Only a few microvilli are seen on the luminal surface and a few mitochondria in the cytoplasm. Some cells have prominent cilia, and different types of cells are found in some cysts. Occasional infoldings of the plasma membranes may be found on the basal surface. The basement membrane of most cysts is strikingly abnormal. There is pronounced splitting, duplication, thickening, and lamination of the basal lamina.
The osmolality of cyst fluids is similar to that of plasma, but sodium and nonsodium osmolyte concentrations vary significantly.
45,
46,
47 Sodium concentrations can vary between 3 and 207 mEq per liter, but often are either less than 60 mEq per liter or more than 75 mEq per liter.
46 Therefore, a distinction was made between low sodium and high sodium cysts. The high sodium cysts have sodium concentrations similar to plasma and therefore are also called nongradient cysts, whereas the low sodium cysts are gradient cysts because they are able to maintain steep concentration gradients not only for sodium but for protons, potassium, chloride, phosphates, and other ions.
48 Morphologically, the gradient cysts have long tight junctions (zonulae occludens depth >500 m), making them impermeable to ions, whereas the nongradient cysts have short tight junctions (<500 m), making them leaky for solutes and water.
46 These characteristics suggested that gradient cysts were derived from collecting ducts and nongradient cysts from proximal tubules. However, most nongradient cysts are lined by a poorly differentiated epithelium with few microvilli and few mitochondria, which does not resemble the proximal tubular epithelium.
46More recent studies have assessed the distribution of aquaporin-1 and -2 in ADPKD cysts. In normal kidneys, aquaporin-1 is expressed in proximal tubules and thin descending limbs of the Henle loop, whereas aquaporin-2 is expressed on the apical surfaces of the collecting duct epithelia. In ADPKD kidneys, approximately 30% of cysts stain positive for aquaporin-1, another 30% are positive for aquaporin-2, and the rest are negative for both aquaporin-1 and -2.
49,
50 The aquaporin-1-positive cysts presumably are derived from proximal tubules or thin descending limbs, the aquaporin-2-positive cysts are from collecting ducts, and the negative cysts are from nephron segments that do not express these water channels (i.e., the ascending limb of the Henle loop and distal convoluted tubules). These results also imply that the expression of water channels is not a prerequisite for cyst expansion. Moreover, the aquaporins appear to retain their segment-specific expression in ADPKD even though the morphologic characteristics of proximal and distal tubules are lost.
In addition to electrolytes and water, cyst fluids also contain amino acids, glucose, urea; idiogenic osmoles such as sorbitol, betaine, and glycerophosphorylcholine; and proteins, such as β
2 -microglobulin, erythropoietin, renin, and albumin.
47,
48 Cytokines, specifically interleukin (IL)-1 β, IL-2, and tumor necrosis factor (TNF)-α, growth factors (epidermal growth factor [EGF], hepatocyte growth factor, endothelins), and a nonpolar lipid cyst activating factor, are present in cyst fluids as well and likely play a significant role in the pathophysiology of ADPKD.
51,
52,
53,
54,
55,
56Therefore, the cysts of polycystic kidneys are not simply impermeable cul-de-sacs that collect and store urine from more proximal nephron segments. They are complex structures that proliferate and undergo apoptosis; that synthesize or transport various proteins, hormones, and cytokines; and that actively secrete chloride and water. Under certain conditions, they also may be able to absorb solutes and water.
45 Most cysts are permeable to small solutes, but some are highly impermeable. Although most cysts are lined by morphologically undifferentiated epithelium, the differential expression of water channels is maintained.
CLINICAL CHARACTERISTICS OF AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE
Although ADPKD derives its name from the kidney, it is a systemic disorder with extrarenal manifestations that are unique to ADPKD.
3,
57,
58 Renal manifestations of ADPKD are
the most common and include the presence of renal cysts, renal enlargement, a decreased renal concentrating ability, polyuria, nocturia, and increased thirst.
59,
60 In addition, renal complications of ADPKD include flank or abdominal pain, gross hematuria, hypertension, urinary tract infections, and nephrolithiasis.
13,
61,
62 Extrarenal manifestations are common in ADPKD and involve the cardiovascular, gastrointestinal, male and female reproductive systems, and the thyroid, subarachnoid, pericardial, and bronchial spaces.
63,
64,
65,
66,
67Although ADPKD is a hereditary renal disease, patients are relatively oligosymptomatic until the second or third decade of life. Renal complications are the most common and occur with increasing frequency with age. Pain is the most common complication, followed by hypertension and gross hematuria. The age-dependent presentation of these manifestations relate closely to kidney size or total kidney volume.
62 By the third decade of life, less than 10% of all ADPKD patients are complication free, even though the mean age of diagnosis of ADPKD is 27 years.
13 Patient-centered outcome reporting demonstrates that thirst, pain, and urinary frequency are the most common patient concerns.
68 However, close attention to other renal complications is important, given their contribution to progressive renal insufficiency and ESRD. Not surprisingly, renal complications are associated with poorer renal and patient outcomes.
Pain is the most common clinical manifestation of ADPKD and is responsible for the majority of presentations for symptomatic diagnosis.
69,
70 Pre-ESRD patients completing quality of life questionnaires demonstrate lower scores on the physical component summary suggesting that symptoms of discomfort significantly impact their quality of life. Focus groups determining the most common patient-reported outcomes find that pain along with thirst and polyuria are the most important. Pain can be managed effectively in most patients, but in a minority, chronic pain limits individuals’ ability to function, resulting in sleep deprivation, fatigue, anxiety, and a decreased quality of life.
71 Acute and chronic pain in ADPKD is due to different causes. Acute pain syndromes in ADPKD are most often associated with cyst rupture, hemorrhage, renal infections, or nephrolithiasis. Chronic pain is a more complex and less defined problem. ADPKD patients report pain located in the back (71%), abdomen (61%), head (49%), chest (30%), and legs (27%).
72 Renal and nonrenal sources of pain not related to cystic disease should be considered including diverticulitis, ovarian cyst rupture, aortic or iliac aneurysms, or incarcerated hernias.
Chronic pain management related to polycystic kidneys requires a staged approach beginning with nonpharmacologic interventions including ice, heat, whirlpool, massage, and physical therapy as well as exercises to improve vertebral and abdominal wall support. When these approaches are not successful, other therapies including intermittent transcutaneous electrical nerve stimulation (TENS) unit and nonopioid analgesics, such as acetaminophen, can improve the level of pain in polycystic patients. There is less objective information regarding the benefits of other treatments including short- and long-term opioid medications, tramadol (Ultram), clonidine, gabapentin, or pregabalin. Nontraditional complementary medical approaches such as acupuncture may be helpful, although this may involve a placebo effect. Surgical approaches to pain in ADPKD patients are reserved for those who have systematically attempted all nonmedical and medical therapies over a reasonable period of time. The least invasive approach is percutaneous cyst aspiration with alcohol injection, typically done in patients with symptoms that can be matched locally to candidate cysts identified using CT or MRI. This can be done in interventional radiology suites as an outpatient procedure. Multiple cyst fenestrations or deroofing procedures (Rovsing procedures) can be done in more severe and complicated cases. Prospective studies report an immediate improvement in 85% to 90% of individuals with close to two thirds maintaining a benefit up to 2 years after treatment.
73,
74,
75,
76 More recently, renal denervation procedures both abdominally and thoracoscopically with and without nephropexy have demonstrated early short-term pain relief.
77,
78 Whether there are long-term benefits resulting from these interventions is not yet clear. Finally, partial or full nephrectomy or volume reducing procedures, including transcatheter arterial embolization, have been used with success in small numbers of patients with intractable pain.
Hypertension
Hypertension is common in ADPKD and, unlike in other tubulointerstitial diseases, it occurs in the majority of patients prior to the loss of kidney function.
79,
80,
81 The average age of onset is 29 years, and men are more often hypertensive than women early in the course of disease.
11,
80 Evidence suggests that carotid and left and right ventricular structure are abnormal in asymptomatic ADPKD patients early in the course of disease prior to the development of hypertension. This is manifest by increases in carotid intimal wall thickness, reduced end-diastolic relaxation, decreased aortic relaxation, increased left ventricular mass, and left ventricular hypertrophy.
82,
83,
84 Hypertension is common in children with ADPKD, affecting 10% to 25% of individuals,
85,
86,
87 and it is associated with evidence of end-organ damage including increased left ventricular mass index and left ventricular hypertrophy. Whether primary cardiovascular abnormalities or increased systemic blood pressure are the primary hemodynamic abnormality to develop in ADPKD and how they relate to each other is unclear. A recent study suggested that an early intervention in ADPKD, particularly with angiotensin-converting enzyme inhibitors or angiotensin receptor blockers, may decrease the occurrence of left ventricular hypertrophy and has the potential to decrease cardiovascular mortality.
88Total kidney volume (TKV) is greater in hypertensive ADPKD adults and children with normal kidney function when compared to the respective normotensive ADPKD controls.
89,
90,
91 The increased cyst burden found in hypertensive ADPKD individuals is associated with evidence of systemic activation of the RAAS. Vascular imaging and MR-based quantification of renal blood flow demonstrate attenuated renal vasculature and reduced renal blood flow,
both of which occur early and are associated with hypertension and increased TKV.
92,
93 Activation of the RAAS in ADPKD is most likely due to increased intrarenal ischemia secondary to cyst formation and expansion. Distortion of the renal vasculature due to increased cyst burden results in decreased renal nitric oxide production, increased reactive oxygen species formation, and further activation of the intrarenal RAAS.
94 Currently two large randomized clinical trials are under way to determine the impact of inhibition of the RAAS and the value of rigorous blood pressure control in disease progression in ADPKD.
95 Results of these trials will be available in 2014.
Gross Hematuria
Gross hematuria is a common initial clinical presentation in ADPKD, often presenting prior to the onset of hypertension. Although not prospectively established, gross hematuria tends to associate with rapid cyst expansion, increased physical activity, and cyst wall calcifications. Gross hematuria is significantly associated with increased kidney size and a poorer renal prognosis.
62,
96,
97 Gross hematuria may or may not be associated with renal pain. In patients with gross hematuria who are asymptomatic, cyst rupture into the urinary collecting system is the most likely cause. Renal imaging is encouraged to rule out other treatable causes of gross hematuria. In patients in whom symptoms develop, localized pain, fever, and dysuria are common. When these clinical signs and symptoms occur, it is important to rule out cyst infection, nephrolithiasis, pyelonephritis, or lower urinary tract infections.
Gross hematuria secondary to cyst rupture is typically self-limited and usually lasts 2 to 5 days. Increased hydration with oral fluid intake and bed rest with close monitoring of blood pressure is indicated given the increased risk for acute reversible kidney injury in the setting of antihypertensive medication intake, particularly angiotensin-receptor blocking agents and angiotensin-converting enzyme inhibitors.
98 Often, it is advised to temporarily discontinue these agents until the episode of gross hematuria resolves. It is important to monitor blood pressure closely with home blood pressure monitoring devices during this time.
Urinary Tract Infections
Urinary tract infections are common in ADPKD and occur more often in women as compared to men.
99 Unlike hypertension and gross hematuria, it is not well established whether urinary tract infections are associated with progressive renal injury. Given that lower urinary tract infections are relatively common in the general population, their occurrence in ADPKD may or may not be disease related. Differentiation between lower and upper urinary tract infections can be difficult, further complicating the discovery of any link between urinary tract infections and disease severity. Upper urinary tract infections in ADPKD may be due to cyst infections, nephrolithiasis, or pyelonephritis and require careful evaluation.
100,
101 Therapy for cyst infections requires different antibiotic treatment than those recommended to treat pyelonephritis in the general population. Antibiotics that provide adequate cyst fluid concentrations are necessary.
102 In addition, a more prolonged course of therapy is needed to ensure successful eradication of the infection. Current recommendations for the treatment of cyst infections include a 2-week course of an oral quinolone or possibly trimethoprim-sulfamethoxazole.
The diagnosis of a cyst infection is often difficult. Clinical presentations vary ranging from local tenderness, fever, leukocytosis, and leukocyturia with positive urine cultures to diffuse abdominal discomfort or pain, absence of a fever, and negative urine cultures.
103 Importantly, blood cultures may more often provide evidence of the infecting organism than urine cultures given that many infected cysts do not directly communicate with the urinary collecting system. Occasionally, cyst infections do not respond to appropriate oral antibiotic therapy. Typically, this occurs in larger cysts (> 5 cm in diameter) or when intracystic antibiotic levels are inadequate. It may be necessary to administer parenteral antibiotics, conduct imaging studies to rule out other causes of fever and pain (including nephrolithiasis), and to consider percutaneous cyst aspiration to obtain cultures or to decompress the large infected space.
104 In rare circumstances, frank pyonephritis may develop associated with sever malaise, sepsis, and shock and may necessitate partial or total nephrectomy.
Nephrolithiasis
Kidney stones are tenfold more common in ADPKD patients than the general population, occurring in approximately 25% of affected individuals.
105 Symptomatic nephrolithiasis typically occurs later than other renal complications in ADPKD.
96 Nephrolithiasis associates with increased TKV in ADPKD patients with normal kidney function. All types of kidney stones can occur in ADPKD; however, urate nephrolithiasis is more common than other types of kidney stones.
106 ADPKD patients develop hypocitraturia, even prior to a loss of renal function, and this may contribute to the increased frequency of urate nephrolithiasis. Whether the hypocitraturia associated with ADPKD is due to abnormalities in renal ammonia generation or other tubular defects is unknown. Of note, urinary biochemical parameters in ADPKD patients uniquely demonstrate normal urinary calcium excretion with increased oxaluria. Patients with nephrolithiasis typically present with unilateral flank pain, with or without radiation, and may have micro- (or rarely, macro-) hematuria. Those with nephrolithiasis diagnosed incidentally during renal imaging more commonly report lower unilateral back pain. Importantly, fevers and chills may occur in the setting of nephrolithiasis. This constellation of signs and symptoms overlap significantly with cyst infections and cyst hemorrhage. An evaluation of nephrolithiasis almost always requires renal imaging—most often, noncontrast CT imaging.
107 Ultrasound has a reduced sensitivity for the detection of kidney stones as compared to CT imaging
in ADPKD individuals. Excretory urography, abdominal flat plate X-rays, and ultrasound can all be used; however, the localization and detection of renal stones is achieved best with CT imaging. The differentiation of renal cyst wall calcification and nephrolithiasis is important and can be easily done when CT imaging is performed. Cyst wall calcifications are more common in ADPKD patients who demonstrate nephrolithiasis than those who do not.
The approach to the management of nephrolithiasis in ADPKD patients should involve a biochemical analysis of the urine and stone using crystallography if possible. Estimates of daily fluid intake and dietary intake of stone-forming elements should be established. Both of these evaluations can be determined from a single 24-hour urine collection. Urinary biochemical analysis should include calcium, urate, oxalate, citrate, and pH.
108 For the majority of stones formed in ADPKD, increases in fluid intake are the cornerstone of therapy. A minimum of 3 L per day of fluid intake should be established using home monitoring coupled with monthly 24-hour urine collections. The addition of bicarbonate or citrate is also helpful for those patients with urate nephrolithiasis to decrease urinary acidification and to increase potential stone dissolution. Dietary education is also helpful with regard to dietary intake of urate, calcium, and oxalate. For patients with calcium oxalate stones, the addition of thiazide diuretics will help to reduce urinary calcium excretion and, coupled with increased fluid intake to greater than 3 L per day, will reduce the concentration of urinary calcium and inhibit initial stone formation.
The most complicated stone in ADPKD patients is the struvite stone, or staghorn calculus. These stones are a constant nidus for infection, which can recur shortly after each treatment course and must be removed to avoid complications. These stones are a nidus for infection, which recurs after each treatment course, and must be removed to avoid complications. Collaboration with the urologic specialists is essential for the management of these patients. Depending of the size of the stone and its location, retrograde lithotripsy, extracorporeal shock wave lithotripsy (EWAL) therapy, or percutaneous stone removal may be necessary.