Fig. 1
COX-2/mPGES-1 pathway. Two primary COX enzyme isoforms (COX-1 and COX-2) catalyze the rate-limiting step of PG formation: the conversion of AA into the intermediate metabolite PGG2, which is further metabolized by different synthases to generate distinct PGs. The specific action of the different PGs in a particular type of tissue predominantly depends on the cell type-specific expression of their specific receptors and on their biosynthesis. NSAIDs (both traditional and COX-2 selective inhibitors, named coxibs) act by the inhibiting COX-dependent prostanoid generation. Innovative anti-inflammatory and anti-cancer strategies are based on the selective delivery of drugs affecting COX-2 activity or expression to specific tissues or by the use of selective inhibitors of mPGES-1, which by suppressing PGE2 might augment the protective vascular PGI2 biosynthesis. In addition, since in CRC COX-2 overexpression is controlled at post-transcriptional level through changes in its mRNA stability by miRNAs, the design of therapeutics affecting this pathway may lead to innovative therapeutic strategies in CRC. AA arachidonic acid, COX cyclooxygenase, PGG 2 Prostaglandin G2, TXA 2 thromboxane, PGI 2 prostacyclin, PGD 2 prostaglandin D2, PGF 2α prostaglandin F2α , TXS TXA2 synthase, PGIS PGI2 synthase, hematopoietic/lipocalin-type PGD synthase; prostaglandin F2a synthase; m(microsomal) or c(cytoplasmatic) PGES (PGE 2 syntase), TP (TXA2 receptor), IP (PGI2 receptor), EP1-4 (PGE2 receptor), DP1-2 (PGD2 receptors), FP (PGF2a receptors), CRC colorectal cancer
NSAIDs, including aspirin, inhibit COX isozymes (COX-1 and COX-2), which catalyze the rate-limiting step in the metabolic conversion of arachidonic acid (AA) to PGs [i.e. PGE2, PGD2, PGF2a, thromboxane (TX) A2, and prostacyclin (PGI2)] (Vane 1971). Prostanoids are second messengers which can cross the cell membrane, diffuse through the extracellular space, and interact with high-affinity G-protein coupled receptors on the same cell or in neighboring cells. The specific action of the different prostanoids in a particular type of tissue predominantly depends on the cell type-specific expression of their specific receptors and on their biosynthesis (Funk 2001) (Fig. 1).
COX-2-derived PGE2 is the most abundant prostanoids found in CRC tissue, and it has been demonstrated to promote colon carcinogenesis along with other malignancies including lung, breast, and head and neck cancer (Wang and Dubois 2010a). The role of PGE2 in colon cancer progression arose from studies with the APCMin/+ mouse model of intestinal neoplasia. This genetic animal model for FAP maintains inactivating mutations in the APC gene (Moser et al. 1995). Treatment of these animals with PGE2 promoted a dramatic increase in small and large intestinal tumor burden (Wang et al. 2004). Moreover, studies in humans revealed that adenoma regression was more effective when PGE2 tissue levels were profoundly inhibited by treatment with NSAIDs (Giardiello et al. 2004). As aforementioned, activation of the canonical Wnt pathway in the colonic epithelium is a key event in polyp formation, and this event is associated with the upregulation of several genes involved in tumor development and progression (Buchanan and DuBois 2006). Among them, overexpression of COX-2 plays a central role in intestinal tumorigenesis. In fact, elevated levels of COX-2-derived PGE2 are associated with (1) resistance to apoptosis, through the upregulation of the anti-apoptotic protein Bcl-2 and the induction of nuclear factor-κB (NFκB) transcriptional activity (Wang and Dubois 2010a), (2) stimulation of cell proliferation, (3) simulation of cell migration, and (4) angiogenesis (Wang et al. 2004).
The well-recognized role of PGE2 during tumor promotion coupled with findings demonstrating long-term use of NSAIDs may be associated with GI toxicity (Masso Gonzalez et al. 2010) and increased risk of adverse CV events (Garcia Rodriguez et al. 2008; Grosser et al. 2006), provided the rationale for the identification of novel enzymatic targets within the AA pathway, including the PGE2 terminal synthases (PGES) (Wang and DuBois 2008). To date, three different gene products with PGES activity have been identified, encoding microsomal PGES-1 (mPGES-1), mPGES-2, and cytosolic PGES (cPGES), respectively (Kudo and Murakami 2005) (Fig. 1). mPGES-1 is a member of the MAPEG (membrane-associated proteins involved in eicosanoid and glutathione metabolism) superfamily, showing significant homology with other MAPEG superfamily proteins. mPGES-1 is expressed at minimal levels in most normal tissues, although abundant and constitutive expression is detected in a limited number of organs, such as the lung, kidney, and reproductive organs. mPGES-1 is also induced by cytokines and various growth factors (Samuelsson et al. 2007). The closely related mPGES-2 is expressed constitutively in a variety of human tissues, and unlike mPGES-1, it is not induced by proinflammatory signals. cPGES is expressed in a ubiquitous manner, and is thought to mediate constitutive PGE2 biosynthesis based on its preferential coupling with COX-1 (Murakami et al. 2000). mPGES-1 is in general functionally coupled with COX-2 and its expression is often concomitantly induced with COX-2 overexpression, thus, contributing to the efficient generation of PGE2 during inflammation (Jakobsson et al. 1999). However, studies using diverse stimuli provided evidence that COX-2 and mPGES-1 can be independently regulated (Sampey et al. 2005). This observation suggested the possibility that the pharmacological targeting of mPGES-1 may result in the suppression of PGE2 production (Fig. 1) by mechanisms that circumvent the CV toxicity associated with inhibition of COX-2 activity by NSAIDs, both traditional and COX-2 selective inhibitors (named coxibs) (Bruno et al. 2010; Wang and DuBois 2008). On the basis of evidence from cell culture studies, several in vivo studies have been performed to address the impact of mPGES-1 targeting on colon tumorigenesis, but the results of these studies are conflicting (Nakanishi et al. 2010). In particular, mPGES-1 knockout mice in a mutant APC background mPGES-1 showed a significant reduction in the number and size of intestinal tumors. Interestingly, mPGES-1 deletion caused a disorganized vascular pattern within primary adenomas, implicating a novel role of PGE2 in tumor angiogenesis (Nakanishi et al. 2008). In contrast, Elander et al. reported that genetic deletion of mPGES-1 resulted in accelerated intestinal tumorigenesis in APCMin/+ mice (Elander et al. 2008). Environmental factors (i.e. Helicobacter pylori infection) may represent a possible explanation of these different responses along with genetic differences in the mouse models used. Based on these differences, the role of mPGES-1 in intestinal carcinogenesis should be further elucidated in preclinical studies prior to extended development of mPGES-1 inhibitors as novel potential chemopreventive agents (Fig. 1) (Nakanishi et al. 2010).
A different mechanism of action seems to explain the chemopreventive effect of long-term use of low-dose aspirin (Rothwell et al. 2010, 2011). In fact, several lines of clinical and experimental evidence support the notion that low-dose aspirin is an efficacious anti-thrombotic agent targeting mainly platelet COX-1 (Patrono et al. 2008; see Chap. 3). Thus, it is proposed that the same mechanism plays a role in the antitumorigenic effect of low-dose aspirin. Activated platelets may be involved in the early phase of intestinal transformation through the activation and induction of COX-2 in stromal cells (Patrono et al. 2001); these cells and their released factors may contribute to epithelial COX-2 expression in the intestinal tract, thus causing the increase of cell proliferation and the accumulation of mutations, as a consequence of inhibition of apoptosis (Cao and Prescott 2002; Prescott 2000). According with this scheme of intestinal tumorigenesis, both COX-1 and COX-2 play a role by operating sequentially, with COX-1 acting upstream of COX-2 (see Chap. 3). This is supported by experimental results showing that loss of either COX-1 or COX-2 genes blocks intestinal polyposis in mouse models of FAP by about 90 % (Chulada et al. 2000). Low-dose aspirin may affect early phase of tumorigenesis by slowing down COX-2 induction in stromal and intestinal epithelial cells, as a consequence of the inhibition of platelet activation. Differently, NSAIDs and coxibs act, as anticancer agents, in later phases of the disease by affecting the consequences of COX-2 overexpression and enhanced generation of prostanoids.
Chan et al. (2007) have shown that regular aspirin use conferred a significant reduction in the risk of CRC that overexpressed COX-2 [multivariate relative risk, 0.64; 95 % confidence interval (CI), 0.52–0.78], whereas regular aspirin use had no influence on tumors with weak or absent expression of COX-2 (multivariate relative risk, 0.96; 95 % CI, 0.73–1.26). These findings do not contradict the antiplatelet mechanism of CRC chemoprevention by aspirin but may suggest that the pathway platelet COX-1 → stromal/epithelial COX-2 overexpression occurs in the setting of genetic/epigenetic dysregulated mechanisms of COX-2 expression.
1.6 Prostaglandin Transporters in CRC
Intracellular PGE2 can cross through the membrane by simple diffusion or via a PG efflux transporter such as MRP4, which efficiently transports PGE2 in an ATP-dependent manner (Reid et al. 2003). Extracellular PGE2 can be transported into the cell, where it can be inactivated or act via nuclear receptors (Zhu et al. 2006). It has been suggested that inactivation of PGE2 in the tumor microenvironment occurs in two steps. First, the PG transporter (PGT) mediates the carrier-mediated membrane transport of PGs, including PGE2, PGF2α , and PGD2 from the extracellular milieu to the cytoplasm. Second, 15-hydroxyprostaglandin dehydrogenase (15-PGDH) catabolizes and thus inactivates PGE2 (Nomura et al. 2004). It has been reported that both the expression and activity of 15-PGDH is repressed in human CRC and APCMin/+ mouse adenomas, resulting in decreased catabolism of PGE2 (Backlund et al. 2005; Myung et al. 2006; Yan et al. 2004). These findings support the role of 15-PGDH as a colon cancer suppressor gene that antagonizes the tumorigenic properties of COX-2-derived PGE2. Recently, this model used to explain the increased levels of PGE2 in colorectal neoplasia has been enriched by novel findings showing that other mechanisms of coordinated up- and downregulation of genes involved in PGE2 transport may contribute to increase PGE2 levels in tumor microenvironment. In particular, it has been shown that PGT and MRP4 mRNA levels are inversely regulated in human CRC compared to normal mucosa and in intestinal adenomas from APCMin/+ mice, thus suggesting that PGE2 transports to the cytoplasm, where it can be inactivated by 15-PGDH may be another crucial step in the regulation of PGE2 levels of intestinal neoplasia (Holla et al. 2008).
1.7 Epidermal Growth Factor Receptor Pathway
Epidermal growth factor receptor (EGFR) mediates another important signaling involving developing colonic carcinomas. In particular, it has been shown that the early effects of COX-2-derived PGE2 are in part mediated by the EGFR. The tyrosine kinase EGFR is one of four members of the HER/ErbB growth factor receptor family and its activity is increased in adenomas that occur in APCMin/+ mice (Moran et al. 2004). Moreover, the disruption of EGFR signaling through either kinase inhibition or genetic mutation inhibits polyp formation and the growth of established tumors (Roberts et al. 2002).
As reported above, PGE2 induces increased proliferation, migration, and invasiveness of colorectal carcinoma cells (Wang et al. 2004). These effects of PGE2 were dependent upon the activation of the phosphatidylinositol 3-kinase/Akt pathway. PGE2 also can induce activation of G proteins which classically activate PKA-mediated transcriptional activation via an increase in cAMP. This coupled activation of Akt and Gα subunits results in the accumulation of β-catenin of the canonical Wnt pathway in the nucleus, leading to transcriptional activation of target genes. Interestingly, it has been shown that the activation of EGFR pathway by PGE2 can also lead to the activation of Akt and MAPK (Buchanan and DuBois 2006). Thus, the transactivation of EGFR by PGE2 is, at least in part, responsible for subsequent downstream effects including the stimulation of cell migration and invasion by COX-2-dependent PGE2. In light of the CV effects associated with long-term COX-2 inhibitor use and the important role of EGFR signaling in CRC, the inhibition of both these pathways has been proposed as a novel potential therapeutic approach and has inspired several studies in animal models of CRC (Buchanan et al. 2007).
1.8 Peroxisome Proliferator-Activated Receptors
Growing evidence has demonstrated that peroxisome proliferator-activated receptor (PPAR) γ serves as a tumor suppressor in CRC. PPARγ is one of the three different isoforms of PPARs (PPARα, PPARβ/δ, and PPARγ), which are ligand-activated transcription factors and members of the nuclear hormone receptor superfamily. They are implicated in a variety of physiologic and pathologic processes such as nutrient metabolism, energy homeostasis, inflammation, and cancer.
Evidence that PPARγ has an antitumor effect in CRC comes from several studies in vitro showing that PPARγ activation is associated with the inhibition of cell growth. This finding was corroborated in animal studies using a xenograft model of CRC in nude mice and another study using mice carrying an heterozygous deletion of PPARγ, where an increased tendency to develop carcinogen-induced colon cancer compared with wild-type mice was observed. Importantly, the protective role of PPARγ is also supported by the finding that point mutations in the ligand-binding domain of one allele of the PPARγ gene resulted in an inability to bind ligands and control gene regulation. Interestingly, despite numerous lines of evidence supporting the role of PPARγ as tumor suppressor gene in CRC, different groups have reported that administration of PPARγ agonists, such as thiazolidinediones, enhanced colon polyp number in the APCMin/+ mice but not in wild-type mice. These results suggest that: (1) PPARγ activation might be associated to a pro-tumorigenic effect when it occurs after tumor initiation, as in the APCMin/+ mice; (2) a predisposed genetic susceptibility is necessary for the occurrence of this pro-tumorigenic effect. Thus, the assessment of the genetic predisposition may be proven useful to predict the success of PPARγ-targeted therapy.
Although it was well established that PPARγ activation is associated with changes of gene expression translating into the induction of apoptosis, inhibition of cell proliferation, and angiogenesis in colon cancer cells, the molecular mechanisms of CRC suppression by PPARγ are complex and not completely elucidated. To date, the main mechanisms implicated in the antitumor activities of PPARγ ligands include the downregulation of β-catenin, the induction of caveolin-1, caveolin-2, and proline oxidase, and the upregulation of the tumor suppressor p53 (Dai and Wang 2010).
1.9 Tumor Microenvironment in Colorectal Carcinogenesis
The tumor microenvironment, which essentially consists of tumor-infiltrating cells, vasculature, extracellular matrix (ECM), and other matrix-associated molecules, has an important role in the multi-step process characteristic of the adenoma-carcinoma sequence in CRC. The tumor microenvironment is quite distinct from the normal tissue microenvironment and it is composed of a particular phenotype of stromal cells, including: (1) tumor-associated macrophages (TAMs), which are activated by microenvironment stimuli to a pro-tumor polarization state (called M2); (2) tumor-associated neutrophils (TANs) with a pro-tumor phenotype (called N2 neutrophils), and cancer-associated fibroblasts (CAFs). In fact, following the initiation of epithelial tumors, reciprocal interactions between transformed epithelial and stromal cells translate into a switch from a normal to a tumor microenvironment which is associated with massive infiltration of immune cells that have undergone substantial changes of their functionality. Tumor-infiltrating cells predominantly include TAMs, myeloid-derived suppressor cells (MDSCs), CD4 T-cells, CD8 T cells, CD4 regulatory T cells (Tregs), mesenchymal stem cells (MSCs), CAFs, endothelial progenitor cells (EPCs), mast cells, and platelets (Peddareddigari et al. 2011). As aforementioned, various reports demonstrate these cells can contribute to elevated prostanoid biosynthesis in the tumor microenvironment through upregulated COX-2 expression (Singer et al. 1998; Wiercinska-Drapalo et al. 1999; Prescott 2000; Cao and Prescott 2002; Dixon et al. 2006) indicating these cells are able to support tumor-associated inflammation, angiogenesis, and immune suppression, which in turn promotes tumor growth and metastasis (Peddareddigari et al. 2011).
2 COX-2 Gene Expression in CRC
As reported above, two primary COX enzyme isoforms have been identified to play distinct roles in physiologic and pathologic conditions. COX-1 is constitutively expressed in most cell types, and COX-1-derived PGs are necessary for protection of gastric mucosa and maintenance of vascular tone (Simmons et al. 2004). While COX-2 is normally absent in most cells, this immediate-early response gene is rapidly induced by a variety of pro-inflammatory and growth-associated stimuli, resulting in increased PGE2 synthesis (Simmons et al. 2004). COX-2 has been identified to play a role in the progression of tumorigenesis, supported by various reports demonstrating that COX-2 levels are increased in premalignant and malignant tumors, and this increase in COX-2 gene expression is associated with decreased cancer patient survival (Wang and Dubois 2010a).
The relationship between COX-2 expression and CRC was first suggested by studies that demonstrated the efficacy of aspirin, NSAIDs, and coxibs to reduce colon cancer risk and also promote tumor regression in both humans and experimental animal models of colon cancer (Wang and Dubois 2010b). The molecular basis of these observations indicated that high levels of COX-2 protein were present in both human and animal colorectal tumors, whereas the normal intestinal mucosa has low-to-undetectable COX-2 expression (Wang and Dubois 2010b). COX-2 is overexpressed in approximately 80 % of colorectal adenocarcinomas and has been shown to play roles in invasiveness, apoptotic resistance, and increased tumor angiogenesis (Wang and Dubois 2010b). This association was further established in genetic studies demonstrating a significant reduction in intestinal polyposis in mice deficient for the COX-2 gene (PTGS2) (Oshima et al. 1996). These findings clearly indicate that chronic elevation of COX-2 is pathological and suggest that inhibition of COX-2 via pharmacological means or regulation of its expression can limit the development and progression of CRC.
2.1 Transcriptional Regulation
A variety of evidence gathered from epidemiological, experimental animal, and cellular studies indicate that unregulated COX-2 expression is an important step in CRC and it is generally well accepted that transcriptional activation of COX-2 can occur early during tumorigenesis (Fig. 2) (Dixon 2003; Wu et al. 2010). Evidence of constitutive activation of the COX-2 promoter occurring in colon cancer cells (Kutchera et al. 1996) suggested that the increased levels of COX-2 mRNA detected in colorectal adenomas and adenocarcinomas (Eberhart et al. 1994; Kutchera et al. 1996) occur through increased transcription. Within the COX-2 promoter region lies cis-acting elements for the binding of several transcriptional factor complexes, such as NFκB (Kojima et al. 2000; Schmedtje et al. 1997; Singer et al. 1998), C/EBP (Kim and Fischer 1998; Shao et al. 2000), β-catenin/TCF (Araki et al. 2003; Howe et al. 1999; Mei et al. 1999), CREB (Shao et al. 2000; Subbaramaiah et al. 2002a), NFAT (Hernandez et al. 2001), AP-1 (Miller et al. 1998; Subbaramaiah et al. 2002b), PPAR (Meade et al. 1999), and HIFα (Bazan and Lukiw 2002; Kaidi et al. 2006). The control of COX-2 transcription is a complex regulatory process that requires input from multiple signal transduction pathways (Dixon 2003). Due to the complexity of combined genetic alterations and inflammatory signaling occurring in the tumor microenvironment, identifying a single transcriptional pathway which plays a decisive role in promoting constitutive COX-2 expression in colon cancer has been limiting. Further efforts involving the molecular characterization of individual tumors will aid in identifying the specific cellular defects in these signaling pathways that can promote aberrant COX-2 gene transcription.
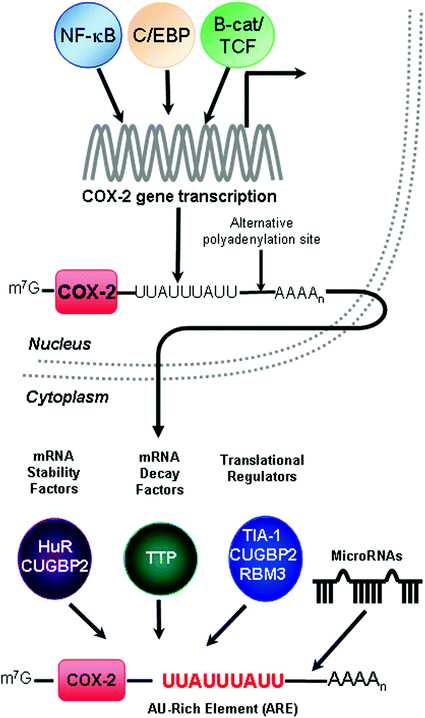
Fig. 2
Regulation of COX-2 gene expression. Transcription of the COX-2 gene PTGS-2 can be induced by various signaling pathways to activate transcription factors NFκB, C/EBP, and β-catenin/TCF. The primary transcript undergoes splicing, capping at the 5′ end, and polyadenylation at the 3′ end; the presence of alternative polyadenylation signals may lead to a shortened 3′UTR and subsequent loss of regulatory elements. After trafficking to the cytoplasm, COX-2 mRNA fate is mediated by sequence elements present within its 3′UTR. COX-2 expression is tightly regulated at the post-transcriptional level by RNA-binding proteins that promote mRNA stability (HuR and CUGBP2), mRNA decay (TTP), and translational inhibition (TIA-1, CUGBP2, and RMB3) through their binding of the COX-2 AU-rich element (ARE). Specific microRNAs have been determined to bind the COX-2 3′UTR and control COX-2 expression
2.2 Post-transcriptional Regulation and the 3′ Untranslated Region
An integral method in controlling gene expression is by post-transcriptional mechanisms that regulate mRNA stability and protein translation. The impact of this level of regulation is evident as microarray analysis has detected that 40–50 % of the changes in inducible gene expression occurs at the level of mRNA stability (Cheadle et al. 2005). Eukaryotic mRNAs contain two characteristic features that are integral to their function, a 5′ 7-methylguanosine cap and a 3′ poly (A) tail. In mammalian cells, the majority of mRNA decay is initiated by shortening of the poly (A) tail (deadenylation), after which degradation initiating at the 5′ end involves removal of the 5′ cap by decapping enzymes, followed by 5′ to 3′ exonucleolytic decay. Alternatively, the mRNA can be degraded by 3′ to 5′ exonucleolytic degradation through a complex of exonucleases known as the exosome (Garneau et al. 2007). These two decay pathways are not mutually exclusive and there appears to be overlap between the pathways, although the relative contribution of each mechanism is under debate (Garneau et al. 2007; Newbury et al. 2006). Many mRNAs targeted for degradation are localized to processing (P)-bodies, which are small cytoplasmic foci that contain components of both 3′ to 5′ and 5′ to 3′ decay machinery, suggesting these decay pathways converge at P-bodies (Eulalio et al. 2007; Garneau et al. 2007). An alternative fate of mRNAs observed under situations of cellular stress is their trafficking to cytoplasmic stress granules where they are translationally silenced. Current work now indicates a functional interaction between P-bodies and stress granules suggesting that mRNAs destined for decay are sorted at stress granules and delivered to P-bodies for degradation (Anderson and Kedersha 2008).
Messenger RNA regulatory elements that play a critical role in identifying specific transcripts for post-transcriptional regulation typically reside within the 3′ untranslated region (3′UTR) of the mRNA (Garneau et al. 2007). A well-established mechanism of 3′UTR-mediated post-transcriptional regulation occurs through association with various RNA-binding proteins that target select mRNAs containing adenylate- and uridylate (AU)-rich elements (AREs) within their 3′UTR (Garneau et al. 2007). More recently, small non-coding RNAs called microRNAs (miRNAs) have emerged as global mediators of post-transcriptional gene regulation through their ability to control mRNA stability and translation by imperfect base pairing to the 3′UTR of its target mRNA (Fabian et al. 2010). Currently, it is estimated that nearly one thousand miRNAs function in humans and have been predicted to regulate approximately 60 % of all protein-coding genes (Friedman et al. 2009).
Identification of multiple mRNA regulatory elements present within the COX-2 3′UTR was the first evidence suggesting that COX-2 might be regulated at a post-transcriptional level (Dixon 2003). Exon 10 of the human PTGS2 gene contains the entire 3′UTR and within this region are multiple polyadenylation signals with two signals that are primarily used to yield transcripts of ~4.6 and 2.8 kb in length (Hall-Pogar et al. 2005). Observed in most cells is the larger 4.6 kb COX-2 transcript that results from processing at a distal canonical (AAUAAA) polyadenylation site. However, a proximal non-canonical polyadenylation signal (AUUAAA) can yield transcripts with shortened 3′UTRs (Hall-Pogar et al. 2005). The significance of this alternative mRNA processing event has been shown in CRC cells as a polyadenylation variant of COX-2 mRNA lacking the distal region of the 3′UTR was selectively stabilized upon cell growth to confluence (Sawaoka et al. 2003). These findings suggest that COX-2 mRNA can escape rapid decay through alternative polyadenylation site usage resulting in deletion of potential 3′UTR regulatory elements and this phenomenon of shortening of mRNA transcripts appears to be a widespread feature occurring in cancer cells (Mayr and Bartel 2009).
2.3 AU-Rich Elements and ARE-Binding Proteins
A characteristic feature controlling the expression of many inflammatory cytokines, growth factors, and proto-oncogenes is their inherent ability to be targeted for rapid mRNA decay. These cancer-associated gene transcripts are unstable due to the presence of a common cis-acting element known as the AU-rich element (ARE) or ARE (Lopez de Silanes et al. 2007). The importance of this particular RNA element is evident, since estimates ranging from 8 to 16 % of human protein-coding genes contain an ARE sequence within their 3′UTR (Bakheet et al. 2006; Gruber et al. 2010). The functional ARE is present within the mRNA 3′UTR and is most often composed of multiple copies of an AUUUA sequence motif clustered together (Bakheet et al. 2006). Within the COX-2 3′UTR, a 116 nucleotide region containing a cluster of 6 AUUUA sequence elements located near the stop codon serves as the functional ARE (Dixon et al. 2000). post-transcriptional regulation has been shown to be dependent upon this ARE since its presence confers rapid decay of a normally stable reporter gene (Dixon et al. 2000). Furthermore, this AU-rich region is highly conserved in both sequence and location among various species of COX-2, implying that ARE function has been evolutionary conserved (Dixon 2003).
Normal cellular growth is associated with rapid decay of ARE-containing mRNAs and targeted mRNA decay is an essential way controlling their pathogenic overexpression. This aspect of COX-2 regulation is observed in non-transformed intestinal epithelial cells where rapid degradation of COX-2 mRNA occurs (t 1/2 ~ 13 min) (Sheng et al. 2000). However, a number of observations have implicated the loss of ARE-mediated post-transcriptional regulation to occur during neoplastic transformation of cells (Lopez de Silanes et al. 2007) and recent findings have demonstrated an enrichment of this subset of transcripts to occur during colon tumorigenesis. Gene expression profiling comparing adenomas to late-stage adenocarcinomas show a three- to fourfold enrichment in ARE-containing genes compared to the genome as a whole and a similar enrichment is observed as early as stage I tumors (Kanies et al. 2008), suggesting loss of ARE-mediated regulation is lost early during tumor development. With regard to COX-2 regulation, similar findings have been observed in human colon carcinoma cells (Dixon et al. 2001, 2003; Shao et al. 2000; Sheng et al. 2000). As a result of the inability of the COX-2 ARE to function properly in CRC cells, enhanced mRNA stability was detected and increased expression of a reporter gene containing the COX-2 3′UTR was also observed. Based upon the inherent genetic instability of tumor cells, it might be expected that mutations or loss of AREs might occur. However, few naturally occurring mutations in AREs have been described and the ARE region of the PTGS2 gene is intact in healthy individuals as well as in colon tumor cells (Dixon 2003). This implies that loss of ARE function in colon tumors is primarily due to altered ARE recognition by cellular trans-acting regulatory RNA binding proteins.
AREs mediate their regulatory function through the association of trans-acting RNA-binding proteins that display high affinity for AREs. The best studied ARE-binding proteins can promote rapid mRNA decay, mRNA stabilization, or translational silencing (Garneau et al. 2007). Through these mechanisms, ARE-binding proteins exhibit pleiotropic effects on gene expression, since a single ARE-binding protein can bind to multiple mRNAs and binding can occur among different classes of AREs (Barreau et al. 2005; Lopez de Silanes et al. 2007). Various cytoplasmic proteins have been detected to bind AREs and work has focused on identifying and characterizing COX-2 ARE-binding proteins. To date, 16 different RNA-binding proteins have been reported to bind the COX-2 3′UTR (Young and Dixon 2010). This chapter section will focus on well-documented RNA-binding proteins that regulate COX-2 expression and their potential impact on CRC (Fig. 2).
2.3.1 HuR
Hu antigen R (HuR; ELAVL1) is a ubiquitously expressed member of the ELAV (Embryonic-Lethal Abnormal Vision in Drosophila) family of RNA-binding proteins (Brennan and Steitz 2001). The human Hu proteins (ubiquitously expressed HuR, and neuronal-specific HuB, HuC, and HuD) were originally discovered as antigens in patients displaying paraneoplastic disorders (Voltz 2002). The cloning and characterization of HuR demonstrated that HuR contains 3 RNA recognition motifs (RRM) with high affinity and specificity for AREs and that cellular HuR overexpression stabilizes ARE-containing transcripts and promotes their translation (Brennan and Steitz 2001).
The ability of HuR to function as an ARE-stability factor appears to be linked to its subcellular localization (Keene 1999). HuR is localized predominantly in the nucleus (>90 %) and can shuttle between the nucleus and cytoplasm. It is hypothesized that the ability of HuR to promote mRNA stabilization requires its translocation to the cytoplasm, and overexpression of HuR promotes cytoplasmic localization where it binds target ARE-containing mRNAs and interferes with their rapid decay (Brennan and Steitz 2001; Keene 1999). A variety of cellular signals known to activate MAPK pathways involving p38 and ERK kinases, the PI-3-kinase pathway, and the Wnt signaling pathway, have been shown to trigger cytoplasmic HuR localization and promote ARE-containing mRNA stabilization (Briata et al. 2003; Ming et al. 2001; Yang et al. 2004). Insight into the mechanism of HuR-mediated mRNA stabilization has been advanced with the identification of low-molecular weight inhibitors for HuR (Meisner et al. 2007). These compounds inhibit HuR cytoplasmic localization by interfering with RNA-binding (Meisner et al. 2007).
HuR has been shown to associate and post-transcriptionally regulate the expression of numerous cancer-associated transcripts bearing AREs of multiple classes (Abdelmohsen and Gorospe 2010; Lopez de Silanes et al. 2005b). Based on its ability to bind the COX-2 ARE, HuR has been identified as a trans-acting factor involved in regulating COX-2 expression (Dixon et al. 2001). The enhanced stabilization of COX-2 mRNA observed in colon cancer cells is, in part, due to elevated levels of HuR (Dixon et al. 2001; Young et al. 2009). Recent work evaluating HuR expression in colon cancer demonstrates that HuR is expressed at low levels and is localized to the nucleus in normal tissue, whereas HuR overexpression and cytoplasmic localization was observed in colon adenomas, adenocarcinomas, and metastases; consistent with these observations, overexpression of COX-2 co-localized with elevated HuR (Young et al. 2009). Furthermore, several studies indicate that HuR overexpression and cytoplasmic localization is a marker for elevated COX-2 that is correlated with advancing stages of malignancy and poor clinical outcome (Dixon 2003).
2.3.2 CUGBP2
CUG triplet repeat–binding protein 2 (CUGBP2) is an ubiquitously expressed RNA-binding protein containing 3 RRM and is a member of the CUGBP-ETR-3-like factors (CELF) family (Mukhopadhyay et al. 2003; Murmu et al. 2004). Work investigating COX-2 regulation in colon cancer cells and intestinal epithelium subjected to ionizing radiation had elucidated a novel role for CUGBP2 in regulating COX-2 expression on a post-transcriptional level (Mukhopadhyay et al. 2003; Murmu et al. 2004). CUGPB2 displays high affinity for the COX-2 ARE and radiation-induced overexpression of CUGBP2 led to COX-2 mRNA stabilization similar to the effects of HuR overexpression (Mukhopadhyay et al. 2003). In contrast to HuR, CUGBP2 repressed COX-2 translation by inhibiting ribosome loading of the COX-2 mRNA (Mukhopadhyay et al. 2003). Given that HuR enhances and CUGBP2 inhibits COX-2 protein expression indicates that these two ARE-binding proteins differ in their regulation of COX-2 expression. Although both proteins have similar affinities for the COX-2 ARE, CUGBP2 was effective in competing with HuR for ARE-binding leading to a translational block in COX-2 expression (Sureban et al. 2007). While demonstrating the ability of CUGBP2 to regulate the opposing functions of COX-2 mRNA stabilization and translational repression, these findings suggest a possible role for CUGBP2 in the early stages of tumorigenesis by counteracting the effects of HuR overexpression in order to repress COX-2 protein synthesis.
2.3.3 TTP
Tristetraprolin (TTP, ZFP36, TIS11) is a member of a small family of tandem Cys3His zinc finger proteins consisting of TTP, ZFP36L1, and ZFP36L2 (Sanduja et al. 2010). TTP acts on a post-transcriptional level to promote rapid decay of ARE-containing mRNAs by direct ARE binding (Carballo et al. 1998). The binding of TTP to AREs targets the transcript for rapid degradation through association with various decay enzymes (Sanduja et al. 2010). In cells, TTP localizes to P-bodies, which suggests that TTP plays a critical role in ARE-mRNA delivery to cytoplasmic sites of mRNA decay (Franks and Lykke-Andersen 2007; Kedersha et al. 2005). TTP is also a target of phosphorylation by various pathways, and this phosphorylation state likely plays a role in mediating TTP function (Sanduja et al. 2010).
Efforts to characterize the function of TTP have primarily focused on its regulation of inflammatory mediators such as TNF-α and GM-CSF (Carballo et al. 2000; Lai et al. 1999). The physiological consequences of TTP loss is evident, as TTP knock-out mice develop multiple inflammatory syndromes resulting from increased inflammatory factors, including COX-2, due to defects in rapid mRNA turnover (Phillips et al. 2004) and recent work has demonstrated COX-2 to be a target of TTP (Sawaoka et al. 2003; Young et al. 2009). With regard to its role in controlling COX-2 expression in colon cancer, TTP expression is low or not apparent in colon cancer cell lines, adenoma, and adenocarcinoma tissue (Young et al. 2009). This is in contrast to normal tissue where TTP was highest in normal epithelium and predominantly localized to the cytoplasm. Furthermore, adenoviral-mediated delivery of TTP to colon cancer cells resulted in downregulation of COX-2 expression coupled with a dramatic reduction in cell growth and proliferation (Young et al. 2009). These findings indicate that the presence of TTP in normal colon epithelium serves in a protective capacity by controlling expression of various inflammatory mediators including COX-2, whereas the loss of TTP expression in tumors allows for HuR to promote the stabilization and translation of COX-2 mRNA.
2.3.4 TIA-1
TIA-1 (T-cell intracellular antigen 1; TIA-1) was originally identified in activated T lymphocytes and is a RNA-binding protein containing 3 RRM motifs with specificity to mRNAs containing short sections of uridylate repeats (Lopez de Silanes et al. 2005a; Tian et al. 1991). Under normal cellular conditions, TIA-1 is predominantly nuclear, and in response to cellular stress translocates to the cytoplasm where it is associated with untranslated mRNAs in cytoplasmic stress granules, implicating a role in translational regulation (Anderson and Kedersha 2008; Kedersha et al. 2005). TIA-1 has been shown to bind the COX-2 ARE and regulate its expression through translational inhibition without impacting COX-2 mRNA turnover (Dixon et al. 2003). However, in colon cancer cells deficiencies in TIA-1 binding to the COX-2 ARE was observed allowing for increased polysome association with the COX-2 mRNA. In vivo, TIA-1 knockout mice maintain elevated levels of COX-2 leading to the development of arthritis (Phillips et al. 2004). These findings implicate TIA-1 as a translational silencer of COX-2 expression, and suggest that loss of TIA-1 function may be a contributing factor promoting enhanced COX-2 levels in cancer and chronic inflammation.
2.3.5 RBM3
RNA-binding motif protein 3 (RBM3) is a translational regulatory factor consisting of a single RRM domain and a glycine-rich region (Dresios et al. 2005). Its role in COX-2 regulation was initially identified through its binding to the AREs in the murine COX-2 3′UTR (Cok and Morrison 2001), and two hybrid screening analysis identified RBM3 to interact with HuR (Anant et al. 2010). Similar to HuR expression in cancer, RBM3 is significantly upregulated in colorectal tumors and overexpression of RBM3 in fibroblasts promoted cell transformation (Sureban et al. 2008), indicating an oncogenic capacity for this RNA-binding protein.
Consistent with its interaction with HuR, RBM3 is also a nucleocytoplasmic shuttling protein that promotes stabilization of COX-2 mRNA through binding to ARE sequences located in the first 60 nts of the COX-2 3′UTR (Sureban et al. 2008). RBM3 protein also promotes translation of COX-2 mRNA through its 3′UTR (Sureban et al. 2008); however, the mechanism underlying this effect and RBM3′s partnership with HuR to promote COX-2 mRNA translation remains to be determined. Through its ability to promote mRNA stability and translation of otherwise rapidly degraded transcripts, RBM3 is being recognized as a contributing factor promoting enhanced COX-2 expression during tumorigenesis.
Related posts:
 Emerging Role for Anti-inflammatory Agents for Chemoprevention
Agents with Anti-lnflammatory Properties in Chemoprevention of Colorectal Neoplasia
Emerging Role for Anti-inflammatory Agents for Chemoprevention
Agents with Anti-lnflammatory Properties in Chemoprevention of Colorectal Neoplasia
 NSAID Targets and Derivatives for Colorectal Cancer Chemoprevention
NSAID Targets and Derivatives for Colorectal Cancer Chemoprevention
 Pharmacology, Toxicity and Efficacy in Cancer Clinical Trials
Pharmacology, Toxicity and Efficacy in Cancer Clinical Trials
 Inheritance and Strategies for Prevention in Populations at High Risk of Colorectal Cancer (CRC)
Inheritance and Strategies for Prevention in Populations at High Risk of Colorectal Cancer (CRC)
 in Prevention of Sporadic Colorectal Cancer: Current Clinical Evidence and Overall Balance of Risks and Benefits
in Prevention of Sporadic Colorectal Cancer: Current Clinical Evidence and Overall Balance of Risks and Benefits
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


