Fig. 1
Chemical structures of coxibs. Rofecoxib and etoricoxib, celecoxib, and valdecoxib are diaryleterocyclic derivatives containing a phenylsulphone and a phenylsulphonamide moiety, respectively. Parecoxib is the water soluble and injectable prodrug of valdecoxib. Differently from the other coxibs, lumiracoxib is a phenyl acetic acid derivative of diclofenac
NSAIDs, one of the most frequently used therapeutic family of drugs worldwide, are a heterogenous group of compounds often chemically unrelated (although most of them are organic acids) (Burke et al. 2006), which act by inhibiting the synthesis of prostanoids, a family of biologically active mediators generated by the activity of COXs (FitzGerald and Patrono 2001; Simmons et al. 2004). NSAIDs are grouped on the basis of pharmacodynamic features, i.e. COX-1/COX-2 selectivity (Capone et al. 2007). This is assessed in vitro and ex vivo (after dosing) using the human whole blood assays (Patrignani et al. 1994; Patrono et al. 1980) which evaluate the effects of drugs on platelet COX-1 and monocyte COX-2 (Fig. 2). Traditional tNSAIDs are a group of drugs which inhibit both COX-1 and COX-2 at therapeutic doses (also called nonselective NSAIDs), while coxibs are selective inhibitors of COX-2.
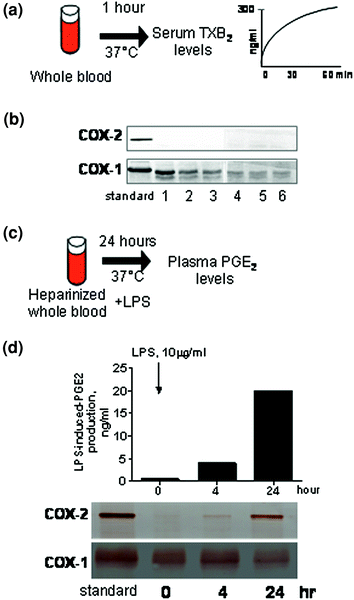
Fig. 2
Whole blood assays to evaluate the effects of COX inhibitors on platelet COX-1 and monocyte COX-2 activities. a The measurement of TXB2 production during whole blood clotting is used as an index of platelet COX-1 activity (Patrono et al. 1980). In panel b, western blot shows that COX-1 but not COX-2 is detected in platelets of healthy subjects (adapted from Patrignani et al. 1999). c The measurement of PGE2 production in response to bacterial endotoxin (LPS) added to heparinized blood samples reflects the time-dependent induction of COX-2 in circulating monocytes (Patrignani et al. 1994). In panel d, the time course of COX-1, COX-2, and PGE2 biosynthesis in monocytes stimulated with LPS is shown. The Western blot of COX-1 and COX-2 in isolated monocytes stimulated (or not) with LPS has been adapted from Sciulli et al. (2003)
tNSAIDs use was its gastrointestinal (GI) safety (Fig. 3). NSAIDs have been repeatedly shown to induce GI events ranging from dyspepsia/gastric intolerance to serious (mainly upper) GI bleeding (Brun and Jones 2001; Hernandez-Diaz and Garcia Rodrìguez 2000). Apart from ASA and NSAID therapy, other factors as advanced age, history of peptic ulcer and comedication with corticosteroids, or anticoagulants have been associated with an increased risk of upper GI disorders (Laine 2001 ; Lanas and Scheiman 2007). Different prevention strategies to overcome/minimize GI problems have been developed in the last decades. The discovery of Helicobacter pylori contribution to the atrophy of the gastric mucosa and subsequently peptic ulcer and gastric cancer onset, together with its eradication therapy have contributed to a better knowledge and control. Some randomized controlled trials (RCTs) and observational studies have found that proton pump inhibitors (PPIs) reduce the risk of upper GI bleeding in patients under antiplatelet and NSAID treatment. Study performed by Lai et al. found that, among ASA users, lansoprazole therapy was associated with a reduced recurrence of ulcer complications when compared with placebo (1.6 vs. 14.8 %) (Lai et al. 2002). Chan et al. showed that among users of NSAIDs other than ASA, omeprazole therapy was associated with a reduced rate of recurrent bleeding compared with Helicobacter pylori eradication therapy (4.4 vs. 18.8 %) (Chan et al. 2001). A recent observational study by Lin et al. observed how PPI use was associated with a lower risk of upper GI bleeding in the general population as well as in patients on antithrombotic or anti-inflammatory therapy (Lin et al. 2011). Thus, according with the ACCF/ACG/AHA 2008 expert consensus guideline concurrent therapy of PPIs is now the standard therapy for patients receiving ASA and NSAIDs (Bhatt et al. 2008).
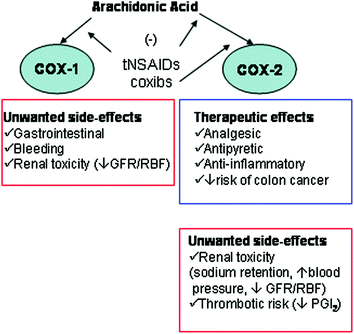
Fig. 3
Pharmacological effects of COX-1 and COX-2 inhibition by traditional (t)NSAIDs and selective COX-2 inhibitors (coxibs). GFR glomerular filtration rate; RBF renal blood flow
Although adverse upper GI events appear to be a class effect, not all tNSAIDs exhibit the same degree of GI toxicity at therapeutic doses. A large body of evidence comparing the GI safety profile across most individual tNSAIDs has been reported in detail (Massó González et al. 2010) and clinicians may now make better-informed clinical and therapeutical decisions on which NSAID to use. With this background of increased risk of GI events observed with all tNSAIDs, the research community together with the industry started to work on the appealing notion of attaining the same analgesic and anti-inflammatory effect, but limiting the undesirable GI effects leading to the development of a novel class of NSAIDs (FitzGerald and Patrono 2001). Inhibition of COX-2 was thought to transduce all the beneficial effects of NSAIDs, while inhibiton of COX-1 was considered to be responsible for all the unintended adverse effects on the GI mucosa (Fig. 3). The introduction of coxibs (Fig. 1) in the 1990s was a major breakthrough thought by many to solve all the GI safety problems once and forever (Box 1). Actually, coxibs demonstrated to be as effective as tNSAIDs and presented an improved GI safety profile though they still carried a small increased risk of serious cardiovascular (CV) events (Grosser et al. 2006). Yet, soon after the market introduction of coxibs which was a huge success, some data started to emerge questioning their effect on CV homeostasis, in particular whether they could increase the risk of acute myocardial infarction (AMI), which led to the withdrawn from the United States (US) and European Union (EU) of two coxibs, rofecoxib and valdecoxib, in 2004, and 2005, respectively. Lumiracoxib was approved in the EU in 2006, but was withdrawn from the market in several countries the following year because of liver toxicity.
An increased risk of hemorrhagic stroke associated with NSAIDs has been also reported in a recent statement by the American Heart Association (AHA) (Antman et al. 2007). Yet, the association between stroke and use of NSAID still remains controversial with several studies yielding conflicting results. A number of studies investigating this relationship have not demonstrated any significant association (Thrift et al. 1999; Bak et al. 2003; Johnsen et al. 2003; Choi et al. 2008). Other authors reported an increased risk of subarachnoid hemorrhage associated with the use of NSAIDs and coxibs (Haag et al. 2008; Roumie et al. 2008; Chang et al. 2010). Use of low-dose ASA has been associated with a small increased risk of intracranial hemorrhage: Antithrombotic Trialists’ (ATT) Collaboration reported a nonsignificant increase (between 30 and 60 %) in hemorrhagic stroke (Baigent et al. 2009). This existing controversy around a true association between NSAIDs and hemorrhagic stroke denotes the necessity of further large-scale studies, especially in target populations as elderly or in a population with multiple potential risk factors for CV events.
This chapter briefly summarizes the current knowledge of chemopreventive role of NSAIDs specially focusing on coxibs on colon cancer onset and progression. Finally, we performed an overview of the different aspects of NSAID-related CV toxicity such as the observed heterogeneity between different individual NSAIDs and its determinants, the time course of the effect, and the potential interaction between ASA and NSAIDs.
2 Box 1
Marketing authorzation was granted for rofecoxib and celecoxib as first representatives of this new pharmacological class in 1999 in the EU and US with the indication for osteoarthritis (OA) and Rheumatoid Arthritis (RA). In 2000, rofecoxib received the marketing authorzation for treatment of acute pain and pain associated with primary dysmenorrhoea in the EU.
During the following years, a number of second generation of coxibs obtained the marketing authorization in the EU and US.
Etoricoxib received the marketing authorization for rheumatic diseases, including gouty arthritis in some EU member states. Valdecoxib was granted the marketing authorization via the EMA central procedure for treatment of RA and OA and pain associated with primary dysmenorrhoea. Parecoxib, the prodrug of valdecoxib, received the marketing authorzation via the central EMA procedure for short-term treatment of post-surgical pain, when used intravenously or intramuscularly. Finally, lumiracoxib received the marketing authorization for symptomatic treatment of OA and acute pain associated with dental and orthopedic surgery and primary dysmenorrhoea. In addition, celecoxib obtained the marketing authorization for an orphan drug indication (FAP) via the EMA central procedure.
From a regulatory point of view, the VIGOR study (Bombardier et al. 2000), together with epidemiological data, which also raised concerns about the CV safety of coxibs, constituted the starting point for a reconsideration of the benefit/risk balance of the coxibs approved at that point of time (celecoxib, etoricoxib, parecoxib, rofecoxib, valdecoxib) with respect to adverse CV and GI effects, in 2002. In addition, serious hypersensitivity and serious skin reactions have been observed with valdecoxib, some in patients with a history of allergic-type reactions to sulphonamides. Thus, an assessment of serious hypersensitivity reactions (e.g. anaphylaxis and angioedema) and serious adverse skin reactions (including Stevens-Johnson syndrome, toxic epidermal necrolysis, erythema multiforme, and exfoliative dermatitis) was added in October 2002, based on concerns raised by epidemiological data. As a result of this first EU coxib-referral, the European Commission concluded in April 2004 that the benefit/risk balance of the coxibs remained favorable; however, that additional warnings should be added to the product informations concerning CV safety (mainly concerning the risk of MI), GI safety (mainly concerning the association with ASA), and observed or potential serious skin effects and hypersensitivity reactions and that the sections on undesirable effects and pharmacodynamic properties should be updated accordingly.
The CV hazard associated with the use of rofecoxib and valdecoxib led to the voluntary withdrawal from the US and EU markets of them, in 2004, and 2005 respectively.
In 2005, FDA has decided to allow celecoxib to remain in the market and has asked Pfizer to revise its label in order to include a boxed warning containing the class NSAID warnings and contraindication about CV and GI risk, plus specific information on the controlled clinical trial data that demonstrate an increased risk of adverse CV events for celecoxib and to encourage practitioners to use the lowest effective dose for the shortest duration consistent with individual patient treatment goals.
Actually, etoricoxib is approved in more than 70 countries worldwide but not in the US, where FDA have required additional safety and efficacy data. Current therapeutic indications are: treatment of RA, psoriatic arthritis, osteoarthritis, ankylosing spondylitis, chronic low back pain, acute pain, and gout. However, the approved indications differ by country.
Valdecoxib was approved by the FDA on 2001 and was available by prescription in tablet form until 2005, when it was removed from the market due to its CV hazard.
Parecoxib is currently available in Europe and is indicated for short-term treatment of postoperative pain, while it has not been approved in US by FDA.
Lumiracoxib is currently marketed in few countries, including Mexico, Ecuador, and the Dominican Republic. In EU, its marketing was approved in 2006, but was withdrawn from the market in several countries the following year because of liver toxicity. It has never been approved for use in the US.
3 Pharmacology of tNSAIDs and Coxibs
Although NSAIDs have been used for centuries, the exact mechanism of action and pathways of these drugs was not unveiled until 1970s (Vane 1971; Simmons et al. 2004). It was discovered that an enzyme called COX is responsible for the crucial step of prostanoid biosynthesis. This enzyme catalyzes the conversion of arachidonic acid into prostaglandin (PG)H2, an intermediate which is converted by different tissue-specific synthases into prostanoids, such as prostacyclin (PGI2), thromboxane (TX)A2, PGE2, PGF2α, PGD2, each with a broad spectrum of biological activities (Simmons et al. 2004; Smyth et al. 2009). There are two isoforms of COX, named COX-1 and COX-2. COX-1 is expressed constitutively in many tissues and it plays a central role in platelet aggregation and gastric cytoprotection (FitzGerald and Patrono 2001), while COX-2 is induced during inflammation, wound healing, and neoplasia. However, COX-2 gene is constitutively expressed in endothelial cells and central nervous system (CNS) (Smyth et al. 2009). As shown in Fig. 3, inhibition of COX-2 by tNSAIDs and coxibs mediates their therapeutic actions (i.e analgesia, antiinflammatory and antitumorigenic effects) but also some unwanted side effects for the CV system. In fact, in endothelial cells of macrocirculation, COX-2 is the major source of PGI2, even in physiological conditions (Grosser et al. 2006; Capone et al. 2007). PGI2 inhibits aggregation of platelets induced by all recognized agonists, vascular smooth muscle cell proliferation and vascular tone, leucocyte-endothelial cell interactions, and cholesteryl ester hydrolase (Grosser et al. 2006). Recently, an antioxidant role for PGI2, through the induction of hemoxygenase-1, has been reported (Grosser et al. 2006; Di Francesco et al. 2009). For all these biological actions, PGI2 has the distinctive features of a cardioprotective mediator. Animal models that were genetically modified for COX-2 or the receptor for PGI2 or TXA2 (named IP and TP, respectively) convincingly showed that reduction of COX-2-dependent PGI2 translates into a hazardous phenotype for the CV system by leaving unconstrained the intricate network of stimuli, such as TXA2, thus predisposing to thrombosis, atherogenesis, and hypertension (Grosser et al. 2006).
Using the human whole blood assays (Fig. 2) which evaluate the effects of drugs on platelet COX-1 and monocyte COX-2 in vitro, the selectivity towards COX-2 by tNSAIDs, and coxibs was characterized (Capone et al. 2007; Patrignani et al. 2008a). In Fig. 4, the COX-2/COX-1 selectivity of the most used NSAIDs is shown. ASA, naproxen and ibuprofen were more potent inhibitors of COX-1 than COX-2, while the majority of NSAIDs (traditional and coxibs) resulted more selective for COX-2 (Capone et al. 2007; Patrignani et al. 2008a). The most selective COX-2 inhibitors are the coxibs rofecoxib, etoricoxib and lumiracoxib, which display COX-1/COX-2 IC50 ratios >100 (IC50: concentration required to inhibit the activity of isozymes by 50 %). However, it was found that COX-2 selectivity is a continuous variable since some tNSAIDs, such as diclofenac, show comparable COX-1/COX-2 IC50 ratios to celecoxib (diclofenac: 24; celecoxib: 30) (García-Rodríguez et al. 2008). Since all these drugs are not specific for COX-2, the degree of COX-2 selectivity obtained in vivo (known as achieved COX-2 selectivity) depends on the dose administered (Capone et al. 2007). In addition to different pharmacodynamic features, NSAIDs are characterized by different pharmacokinetics (PK) parameters, such as half-life, which, by driving the extent and duration of patient drug exposure, are important determinants of their therapeutic and toxic effects in vivo.
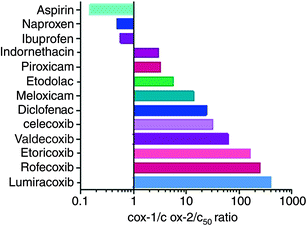
Fig. 4
Biochemical selectivity, assessed as COX-1/COX-2 IC50 values of some COX inhibitors. The value 1 mirrors an equivalent potency to inhibit both COX-isozymes. Higher values (>1) mirror higher selectivity versus COX-2. Lower values (<1) mirror higher selectivity for COX-1
4 Efficacy of Coxibs in CRC Chemoprevention Trials
tNSAIDs and coxibs have shown to be effective in CRC chemoprevention (Cha and Dubois 2007; Wang and Dubois 2010b). A most plausible mechanism involves their shared capacity to inhibit COX-2-dependent PGE2 (reviewed in detail in Chap. 2).
PGE2 has a predominant role in promoting colorectal tumor growth (Wang and Dubois 2010a and b). It is the most abundant PG detected in human CRC (Bennett and Del Tacca 1975; Jaffe 1974; Rigas et al. 1993). PGE2 modulates a number of signal transduction pathways that may affect proliferation, programmed cell death (apoptosis), angiogenesis, immune response, cellular adhesion, differentiation, and tumor invasion (Ferrandez et al. 2003; Wang and Dubois 2010a).
Increased expression of COX-2 probably occurs during all stages of the multistep progression of CRC, from the first genetically altered cell, all throughout the different steps from hyperplasia, dysplasia, to carcinoma, and even metastasis (DuBois et al. 1996; Hao et al. 1999; Shiff and Rigas 1999). Overexpression of COX-2 also increases cell migration and proliferation in intestinal epithelial cells (Koehne and Dubois 2004; Wang and Dubois 2010b). COX-2 has been linked with several premalignant and malignant lesions of epithelial origin in lung, breast, colon, and stomach (Dannenberg et al. 2001). Eberhart and colleagues showed that COX-2 genes are highly elevated in most human CRCs compared with the normal mucosa (Eberhart et al. 1994). Additionally, it has been observed an increased expression of COX-2 in CRC, while COX-1 expression seems to be unaltered (Kargman et al. 1995). This finding was confirmed by other studies that observed an increased COX-2 expression (Rao and Reddy 2004; Sinicrope 2006).
The advanced knowledge towards the association between COX-2 and CRC led many scientists and clinicians to investigate whether the inhibition of this pathway could provide some reduction in CRC risk. Data from several studies have directly and indirectly shown that the anticarcinogenic properties of NSAIDs occur through inhibition of COX-2 but other indirect mechanisms could be also involved (Ferrandez et al. 2003; Baek et al. 2001; Goel et al. 2003; Pan et al. 2008; Jana 2008; Chan 2002; Husain et al. 2002). In fact, it was shown that cells that do not express COX-2 also undergo apoptosis in response to exposure to NSAIDs (Arber 2008). However, these studies were performed in vitro by using higher concentrations than those obtained in vivo. Thus, it seems to be unlikely that these COX-independent mechanisms may be involved in the antitumorigenic effect of NSAIDs detected at therapeutic doses.
The first clinical trial in cancer prevention using the selective COX-2 inhibitor celecoxib was carried out in the setting of familial adenomatous polyposis (FAP) patients with intact colon in 2000 year (Steinbach et al. 2000). Patients with FAP have a nearly 100 % risk of developing CRC. The aim of this trial was to evaluate the chemopreventive effects of celecoxib. This trial was a randomized, double blind, placebo controlled. Patients were randomly selected to put on two different plural, dosages of celecoxib (400 or 100 mg/BID) or placebo for six months. Patients underwent endoscopy at the beginning and end of the trial and the main endpoint was the average polyposis reduction from baseline. Upon completion of the study, a significant reduction in polyp burden (by 30 %) was observed in patients who received 400 mg of celecoxib twice daily whilst around 12–15 % reduction was observed in the group receiving celecoxib with 100 mg/BID and placebo, respectively. Based on these results, the FDA approved the use of celecoxib at 800 mg daily as an oral adjunct therapy for the treatment of patients with FAP in 1999. However, more recently, Pfizer has voluntarily withdrawn the indication for reduction of colorectal polyps in patients with FAP for celecoxib, because it was unable to provide confirmatory data regarding clinical benefit due to slow enrolment in an ongoing clinical trial.
Another trial performed with rofecoxib and a mean follow up of 16 months, focused on assessing the maintenance of colon free of polyps in FAP. The trial encompassed only eight patients who were put on rofecoxib (25 mg/day) for 30 months and sigmoidoscopy/colonoscopy was done at entry and every six months. The number, size, and histologic grade of all polyps were assessed, and the polyps were removed during each endoscopic procedure. The efficacy of the combined approach of endoscopy and chemoprevention was shown with a highly significant reduction in the rate of polyp formation (by 70–100 %) at the end of the study. The investigators concluded that long-term use of rofecoxib was well tolerated and effective in inhibiting polyp formation in polyposis patients (Hallak et al. 2003).
In addition, three studies were carried out to examine the efficacy and safety of coxibs in preventing the recurrence of sporadic colorectal polyps. The design of these trials was multi-center, prospective, randomized, and placebo-controlled trial studies and all required continuous treatment for approximately three years, with a 2-year extension to evaluate drug safety. Each study recruited between 1,500 and 2,600 patients who had undergone a recent adenoma removal. The Adenomatous Polyp Prevention On Vioxx (APPROVe) trial was a randomized, double blind, placebo-controlled trial of the efficacy of oral rofecoxib, 25 mg/day, to prevent colorectal adenomas (Baron et al. 2006; Bresalier et al. 2005). For regulatory purposes, the main aim of the study was a three years trial with rofecoxib among subjects at high risk of developing adenomas, meeting the following criteria: having an adenoma 1 cm or greater in diameter, an adenoma with villous or tubulovillous histology, two or more adenomas, younger than 55 years at first adenoma diagnosis and/or history of colon cancer among first degree relatives. The study recruited a total of 2,586 patients. Participants were assigned to receive rofecoxib 25 mg daily (1,257 patients) or placebo (1,299 patients). The authors found that rofecoxib significantly reduce the risk of recurrent adenomas among patients with a recent adenoma history. However, the study was terminated a few months before the planned end of the trial following the advice of the External Safety and Monitoring Board because of a higher rate of CV events in the rofecoxib group.
The Adenoma Prevention with Celecoxib (APC) trial was a randomized, placebo-controlled trial that investigated whether celecoxib reduces the occurrence of endoscopically detected colorectal adenomas (Bertagnolli et al. 2006). This trial included 2,035 randomized patients with a recently removed adenomatous polyp. They were at high risk of recurrent adenomas (e.g., based on a history of either multiple adenomas or removal of a single adenoma more than 5 mm in diameter), and were randomized to either placebo or celecoxib (200 or 400 mg/BID). These patients were followed up for a mean of 33 months while on treatment. The cumulative incidence of detection of one or more adenomas by year 3 was 60.7 % in patients receiving placebo versus 43.2 % for those receiving celecoxib 200 mg/BID (risk ratio, 0.67; 95 % CI, 0.59–0.77; P < 0.001) and 37.5 % for those receiving 400 mg of celecoxib twice a day (risk ratio, 0.55; 95 % CI, 0.48–0.64; P < 0.001). These authors concluded that celecoxib was an effective agent for the prevention of colorectal adenomas but, because of potential CV events, could not be routinely recommended for this indication.
The Prevention of Sporadic Adenomatous Polyps (PreSAP) trial was also a randomized, placebo controlled, double-blind study of the COX-2 inhibitor celecoxib given daily in a single 400 mg dose conducted in parallel to the APC trial for the same indication: 1,561 patients from 107 medical centers in 32 countries from six continents were randomized (3:2) to receive either celecoxib (400 mg) or placebo. Celecoxib was associated with a relative risk (RR) of 0.64 for adenomas detected during a 3-year period. A reduced risk was already apparent at the first year follow-up colonoscopy. The adenoma recurrence rate was 33 % in the celecoxib group versus 49.3 % in the placebo group (P < 0.0001) (Arber et al. 2006).
All the results from these cancer prevention trials with coxibs generated major expectations. However, many concerns on their CV safety arose at the same time (Solomon et al. 2005). In 2004, rofecoxib was withdrawn unilaterally by Merck from the market due to increased CV toxicity observed in APPROVe (Baron et al. 2006; Bresalier et al. 2005). Also, a few months later the FDA issued a “black box” warning for valdecoxib (Bextra, Pfizer) due to increased CV risk in patients undergoing coronary artery bypass (Nussmeier et al. 2005).
With regards to observational epidemiological data, few studies have investigated the link between exposure to coxibs on colorectal occurrence and recurrence. A nested case-control analysis carried out by Rhame et al. showed that exposure to at least three months of rofecoxib or nonselective NSAIDs (all doses) had a significant protective effect, also these authors found a trend towards a greater reduced risk with high-dose than low doses (Rhame et al. 2002).
5 CV Toxicity of Coxibs
5.1 Risk Estimates: Data from Trials and Observational Studies
Some of the first coxib trials were focused in quantifying the expected improved safety profile (mainly on GI safety) among long-term users of these drugs compared to selected tNSAIDs with an average follow-up duration of one year. The control group consisted of naproxen, ibuprofen or diclofenac and/or placebo if possible, but for some instances like RA patients, the use of placebo was not an option. The first study showing an increased risk of myocardial infarction (MI) associated with a coxib was the VIGOR trial. This trial was initially designed to compare the GI safety of rofecoxib (50 mg/day) and naproxen (500 mg/BID) in patients with RA. Although rofecoxib demonstrated a lower risk of GI events, the finding of a 4 to 5-fold increased risk of MI among users of rofecoxib marked an inflection bent in the assessment of the safety profile of these drugs (Bombardier et al. 2000). However, this effect was mainly described as not related to an increased risk of rofecoxib itself, but to an intrinsic and previously unnoticed major cardio-preventive effect of naproxen (Bombardier et al. 2000). This hypothesis carried on for quite some time despite the following two facts. Firstly, though naproxen at high doses certainly confers a profound and persistent inhibition of platelet COX-1 (Capone et al. 2004), due to its long half-life (i.e 17 h) (Burke et al. 2006), this blockade, unlike the one produced by low-dose ASA, which affects irreversibly COX-1, is time dependent and reversible. Secondly, even in the unlikely event that naproxen would share the same beneficial effect than ASA (which is estimated to reduce the risk in secondary prevention between 20 and 30 %) (Patrono et al. 2008), it could never explain a five-fold increased risk observed among rofecoxib users compared to naproxen users (Bombardier et al. 2000). An increasing number of observational studies and several RCTs analyzing the CV risk profile of coxibs and some tNSAIDs made their appearance over the next decade after the VIGOR results were first published. Eventually, some years along the road all NSAIDs, including tNSAIDs and not just coxibs, were shown to increase, to different degrees, the risk of ischemic CV events and, in particular, of AMI.
The overall results of CLASS, a parallel study performed to compare GI safety of celecoxib (400 mg/BID), diclofenac (75 mg/BID), and ibuprofen (800 mg/TID) failed to detect a significant difference in GI or CV events between various treatment arms (Silverstein et al. 2000). This study was subject to many methodological criticisms that could explain in part these results. The TARGET trial (that was actually comprised by two sub-studies) recruited around 18,000 osteoarthritis patients randomized to lumiracoxib (400 mg), naproxen (500 mg/BID), or ibuprofen (800 mg/TID) during one year (Farkouh et al. 2004). Overall, this study was unable to find significant differences in MI risk neither between lumiracoxib and ibuprofen (RR, 95 % CI: 0.66, 0.21–2.09) nor between lumiracoxib and naproxen (RR, 95 % CI: 1.77, 0.82–3.84). It should be noted that these results were based on a limited number of events (a total of 33 cases) (Farkouh et al. 2004). Some other clinical trials were designed with the main objective to study a reduction in the risk of CRC. However, these trials reported an increased CV risk associated to coxibs and some had an early termination.
APPROVe was a study with three years of planned follow up, assessing as primary endpoint the recurrence of adenomatous polyps in patients with antecedents of colorectal adenomas in patients receiving rofecoxib (25 mg/day) or placebo (Baron et al. 2006). The study was terminated prematurely due to elevated incidence of CV events in the rofecoxib arm (RR: 1.92, 1.19–3.11) (Bresalier et al. 2005). Two similar studies were conducted for celecoxib, the APC, and the PreSAP comparing varying doses of celecoxib (APC: 200 mg/BID and 400 mg/BID; Pre-SAP: 400 mg/daily) with placebo (Solomon et al. 2005; Solomon et al. 2006a). Some preliminary results of APC, suggesting a dose-related two-fold increased CV mortality of celecoxib compared to placebo and this motivated the termination of APC when the study was close to be completed. Additionally, a meta-analysis including these and other RCTs not considered here with either shorter follow up, smaller sample sizes, or both, estimated that coxibs were associated with a 42 % increased risk of serious vascular events compared to placebo (RR, 95 % CI:1.42, 1.13–1.78) (Kearney et al. 2006).
Among observational studies assessing NSAIDs and CV risk, most of them were conducted in large cohorts using automated databases as the primary source of information and only a small number were hospital-based case-control studies.
These studies had several strengths like the ability to identify population-based controls from the underlying study cohort, large sample sizes, and the absence of recall bias since exposure was ascertained prospectively (i.e. before the event actually occurred). However, it should be noted that most of these studies had the limitation of not being able to capture over-the-counter (OTC) drug use. However, there is no reason to believe that OTC use was more common in cases than controls. The study performed by Ilkanoff et al. studied this issue and estimated that excluding users of OTC NSAIDs from the unexposed group would result in around 10 % change away from the null (Ilkhanoff et al. 2005).
Among the few observational studies that did not use automated databases, two studies were conducted in a network of 36 hospitals in the Philadelphia area (Kimmel et al. 2004, 2005) and a reanalysis of data from a clinical trial, the physician health study (PHS) (1989), carried out in the 1980s to evaluate the overall efficacy of ASA in reducing the incidence of primary MI. Most studies analyzed first MI or first MI hospitalization as the main endpoint, but there were some studies that either did not exclude cases with antecedents of MI prior to the study period (Kimmel et al. 2005; Ray et al. 2002) or specifically followed patients from a first MI to a second CV event or death (Gislason et al. 2006; Curtis et al. 2003; MacDonald and Wei 2003).
Three different meta analyses have been published to date summarizing the results from observational studies (Kimmel et al. 2004, 2005; Mamdani et al. 2003; García Rodríguez et al. 2004; Solomon et al. 2004; Graham et al. 2005; McGettigan et al. 2006; Singh et al. 2005, 2006; Sturkenboom et al. 2005; Gislason et al. 2006; Curtis et al. 2003; MacDonald and Wei 2003; Hernandez-Diaz et al. 2006; Singh et al. 2006; McGettigan and Henry 2006; Kurth et al. 2003; Ray et al. 2002; Solomon et al. 2002; Watson et al. 2002; Schlienger et al. 2002; Lévesque et al. 2005; Johnsen et al. 2005; Hippisley-Cox and Coupland 2005; Fischer et al. 2004; Bak et al. 2003). Among them, the RR (95 % CI) associated to tNSAIDs ranges from 1.08 (0.95–1.22) to 1.19 (1.08–1.31). Some observational studies were not included in these meta analyses (García-Rodríguez et al. 2008; Andersohn et al. 2006; Varas-Lorenzo et al. 2009; Velentgas et al. 2006; Suissa et al. 2006; Ray et al. 2009; Solomon et al. 2006b; Helin-Salmivaara et al. 2006; Bueno et al. 2010). One of them had a very large sample size. This is a nation-wide Finnish study of discharge summaries and included a total of 33,309 incident cases of MI (Helin-Salmivaara et al. 2006). Overall the results of this study are congruent with previously detailed summarized data. The risk of MI associated with individual NSAIDs went from a 1.06 (0.83–1.34) for celecoxib to 2.21 (1.18–4.14) for etoricoxib.
In APPROVe trial, time to event associated with coxib use compared to placebo suggested that the deleterious effect appeared only after 18 months of initiating treatment (Bresalier et al. 2005). This finding generated a great deal of controversy, because previous studies with shorter follow up were also successfully able to detect an increased risk (Bombardier et al. 2000). New data from an extended follow-up of the APPROVe study was lately published (Baron et al. 2008), and the authors reported that the increased CV risk observed among individuals exposed to rofecoxib persisted for some time after discontinuation: the RR was 1.95 (0.97–3.93) in the first year after discontinuation. To confirm this finding, we used data from a nested case-control study performed by our group, and identified individuals who discontinued NSAID use between 7 and 365 days before the study index date (García-Rodríguez et al. 2008). Overall we found that among those discontinuing NSAID recently, long-term NSAID users for one year or more, RR was 1.58 (1.27–1.96). We also found a similar RR of AMI among patients currently exposed to NSAIDs with duration of more than one year (1.45, 1.27–1.65). These results suggest that tNSAIDs could also carry a persisting risk for a limited period of time after treatment discontinuation. Yet, the exact nature between NSAID duration and the CV hazard remains uncertain.
5.2 Mechanisms of CV Toxicity of Coxibs
The most plausible hypothesis of the CV hazard associated with the administration of coxibs is that they affect COX-2-dependent PGI2 generation, while not affecting platelet function (García-Rodríguez et al. 2008; Grosser et al. 2006). In fact, a cardioprotective phenotype can be obtained by the administration of low-dose ASA through its capacity to almost completely (≥95 %) (Reilly and FitzGerald 1987 ) and persistently suppress platelet TXA2 generation and function throughout dosing interval (Patrono et al. 2008), while leaving almost unaffected vascular PGI2 generation. ASA is an irreversible inhibitor of COX-1 and COX-2, through selective acetylation of a specific serine residue of Ser529 and Ser516, respectively (Patrono et al. 2008). However, ASA has a short half-life (approximately 20 min), thus when administered at low doses (75–100 mg) once daily, it preferentially inhibits platelet COX-1 in the presystemic circulation. Since platelets are anucleated cells with limited capacity to synthesize proteins de novo, the irreversible inhibition of COX-1 persists for all platelet lifespan (i.e. 7–10 days). This ability to permanently inhibit TXA2 production in platelets places ASA in a unique position to be used in coronary heart disease prevention (Patrono et al. 2008; see also Chap. 3).
Related posts:
 Emerging Role for Anti-inflammatory Agents for Chemoprevention
Agents with Anti-lnflammatory Properties in Chemoprevention of Colorectal Neoplasia
Emerging Role for Anti-inflammatory Agents for Chemoprevention
Agents with Anti-lnflammatory Properties in Chemoprevention of Colorectal Neoplasia
 Aspects of COX-2 Expression in Colorectal Neoplasia
Aspects of COX-2 Expression in Colorectal Neoplasia
 NSAID Targets and Derivatives for Colorectal Cancer Chemoprevention
NSAID Targets and Derivatives for Colorectal Cancer Chemoprevention
 Inheritance and Strategies for Prevention in Populations at High Risk of Colorectal Cancer (CRC)
Inheritance and Strategies for Prevention in Populations at High Risk of Colorectal Cancer (CRC)
 in Prevention of Sporadic Colorectal Cancer: Current Clinical Evidence and Overall Balance of Risks and Benefits
in Prevention of Sporadic Colorectal Cancer: Current Clinical Evidence and Overall Balance of Risks and Benefits
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


