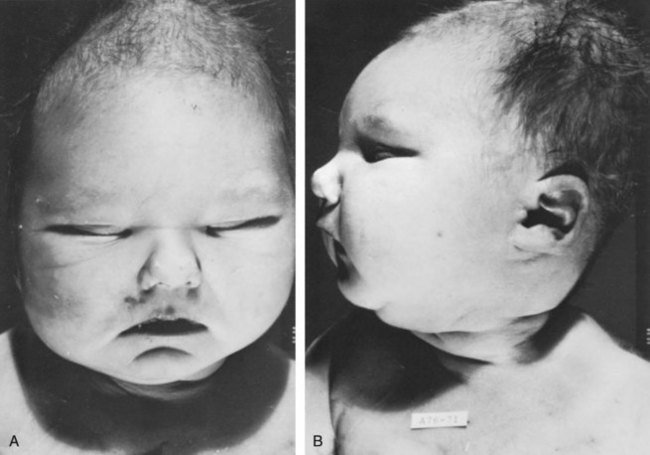
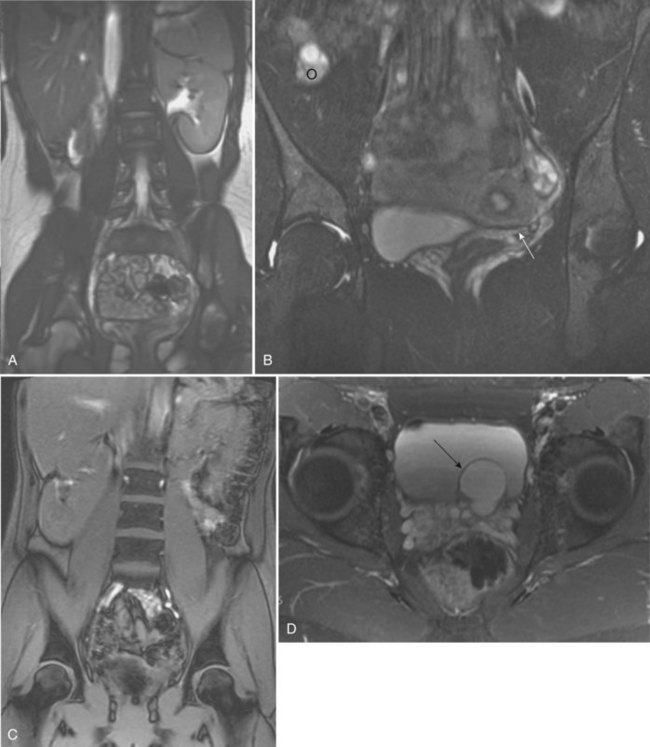
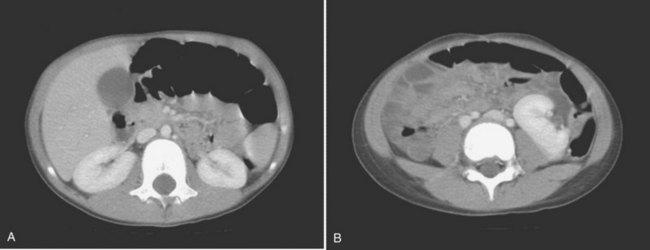
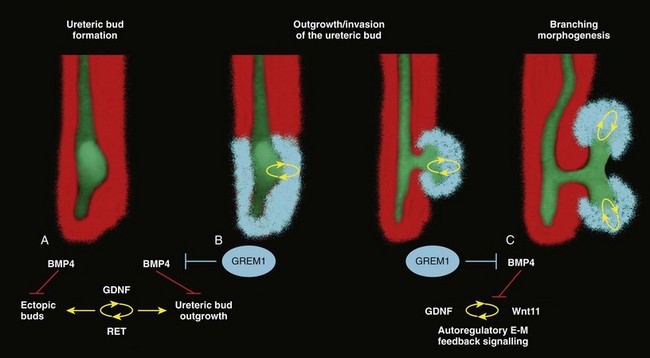
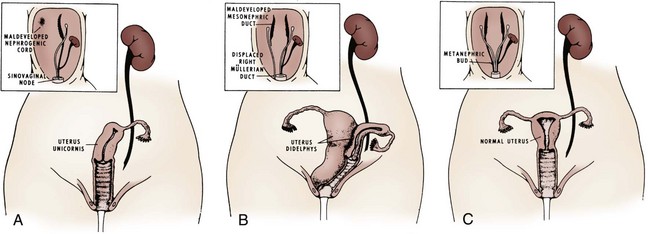
Ellen Shapiro, MD, Stuart B. Bauer, MD, Jeanne S. Chow, MD Of all the anomalies of the upper urinary tract, bilateral renal agenesis (BRA) has the most profound effect on the fetus. Fortunately, it occurs infrequently when compared with other renal abnormalities. Although BRA was first recognized in 1671 by Wolfstrigel, it was not until Potter’s extensive description of the constellation of associated defects that the full extent of the syndrome could be appreciated and easily recognized (Potter, 1946a, 1946b, 1952). Subsequently, many investigators have attempted to explain the syndrome by employing a single unifying etiology (Fitch and Lachance, 1972). However, we are learning that there is probably no single etiology as we begin to unravel the myriad of complex molecular events that are required for normal renal development. The incidence of BRA is rare, with only about 500 cases reported in the literature. Potter (1965) estimated that BRA occurs once in 4800 births, but in British Columbia the incidence is 1 in 10,000 births (Wilson and Baird, 1985). Davidson and Ross (1954) noted a 0.28% incidence in autopsies of infants and children, whereas Stroup and colleagues (1990) detected an incidence of 3.5 per 100,000 in the Centers for Disease Control Birth Defects Monitoring Program. A recent study using prenatal ultrasonography in 8500 pregnancies in Poland reported an incidence of 0.25% (Forys et al, 2003). There is significant male predominance, with almost 75% of individuals being male. Increasing maternal age appears to be a risk factor (Bianca et al, 2003), but specific complications of pregnancy or any maternal disease do not appear to consistently influence the incidence of BRA (Davidson and Ross, 1954; Ruhland et al, 1998). The anomaly was reported in three infants of an insulin-dependent diabetic mother (Novak and Robinson, 1994). It has been observed in several sets of siblings (Rizza and Downing, 1971; Dicker et al, 1984) and in monozygotic twins (Thomas and Smith, 1974; Cilento et al, 1994). In four pairs of monozygotic twins, one sibling was anephric while the other had normal kidneys (Kohler, 1972; Mauer et al, 1974; Cilento et al, 1994; Klinger et al, 1997). It has been suggested that an autosomal-recessive inheritance pattern exists (Dicker et al, 1984). There is a genetic predisposition to this syndrome with a high level of penetrance (Stella, 1998). When siblings and parents of an index child with BRA were screened, 4.5% had unilateral renal agenesis (Roodhooft et al, 1984) and 3.5% had BRA (McPherson et al, 1987). This is 1000 times higher than in the general population (Stroup et al, 1990). Other investigators have suggested that this is an autosomal-dominant trait with variable penetrance (Kovacs et al, 1991; Murugasu et al, 1991; Moerman et al, 1994; Stella, 1998). Recently, McPherson (2007) evaluated renal anomalies in families of individuals with congenital solitary kidneys that included renal agenesis or a very poorly functioning kidney due to dysplasia/hypoplasia. The empiric risk of 7% for offspring, 4% for parents, and 2.5% for siblings may be an underestimation, because not all relatives underwent ultrasound screening. The incidence of BRA in offspring of congenital solitary kidney probands was ≈1%, which is significantly greater than the risk found in the general population but less than that for families with a history of BRA. Ultrasound screening has been recommended for parents and siblings of infants born with either unilateral or bilateral renal agenesis or dysgenesis (Roodhooft, 1984). McPherson (2007) has recommended prenatal ultrasound examination when either parent has a congenital solitary kidney. Ultrasound screening is also recommended for first-degree relatives of persons with congenital solitary kidney. BRA has been detected in higher-than-expected proportions in esophageal atresia (Saing et al, 1998) and several syndromes, including cryptophthalmos or Frazer syndrome (Fryns et al, 1997), Klinefelter syndrome (Barroeta et al, 2004), and Kallmann syndrome (Colquhoun-Kerr et al, 1999). The intermediate kidney, or mesonephros, develops and then regresses except for the mesonephric tubules (Constantini and Shakya, 2006; Schedl, 2007; Uetani and Bouchard, 2009). In the male, these are the efferent ductules that serve as a link between the gonad and the mesonephric or wolffian duct (WD) structures (the body and tail of the epididymis and vas deferens). In the female, the mesonephric tubules link the ovary through the fimbriated end of the fallopian tube to the reproductive tract. The WD elongates caudally and fuses with the anterior cloaca. The definitive kidney differentiates from the metanephric blastema, which is a specialized region of the intermediate mesoderm termed the metanephric mesenchyme (MM). This process requires the reciprocal induction between the metanephric blastema and the ureteral bud (UB). The metanephric blastema sends signals to the WD to initiate UB formation between the fifth and seventh weeks of gestation. As a result, the ureter is induced from the caudal end of the WD. The UB evaginates and invades the metanephric blastema and branches repeatedly in a characteristic pattern to form the collecting duct system. The ureteral tips induce nephron differentiation in the adjacent mesenchyme, forming the mature metanephros (Airik, 2007; Uetani and Bouchard, 2009). The absence of a nephrogenic ridge on the dorsolateral aspect of the coelomic cavity or the failure of a UB to develop from the WD will result in renal agenesis. Therefore in order for BRA to occur, there must be an alteration in normal molecular development or a mutation that causes renal or ureteral maldevelopment on both sides of the midline (see Molecular Mechanisms of Mammalian Kidney Organogenesis). A review of the relationship of the WD to müllerian duct (MD) development is necessary to understand the genitourinary phenotype of individuals with renal anomalies or more specifically, renal agenesis (Kobayashi and Behringer, 2003). The cellular mechanisms involved in MD formation have been partially unraveled only recently. Gene fate mapping and lineage tracing experiments show that the WD does not contribute cells to the MD, and the MDs are derived from the coelomic epithelium (Guioli et al, 2007; Orvis and Behringer, 2007). A three-phase model of MD development has recently been proposed (Guioli et al, 2007; Orvis and Behringer, 2007). In the first phase, cells of the coelomic epithelium in the cervical region of the intermediate mesoderm are specified to become MD cells and have been noted to express Lim1 (Kobayashi et al, 2005; Orvis and Behringer, 2007; Masse et al, 2009) (Fig. 117–1). After the process of specification is complete, the second phase is heralded by Wnt4 expression from the mesonephros or coelomic epithelium, which induces these cells that are destined to become the MDs to invaginate (Kobayashi et al, 2004, 2005; Orvis and Behringer, 2007). The second phase of MD development is WD independent and ends when the MD extends caudally and contacts the WD (Carroll, 2005; Kobayashi et al, 2005; Orvis and Behringer, 2007). The third phase involves elongation of the MDs posteriorly until they are joined at the urogenital sinus. This process is WD dependent, requiring the MD epithelium at its posterior end to be in close physical contact with the WD epithelium, while the MDs are separated from the coelomic epithelium by only a basement membrane (Orvis and Behringer, 2007). This intimate relationship between the WD and MD is emphasized in experiments that interrupt the formation of the WD at a specific point and show that the MD could not grow beyond that point to complete its formation (Gruenwald, 1941). Lim1 in the WD is critical for WD maintainance. Loss of Lim1 in the WD by inactivation leads to WD loss. Because the MD is dependent upon the WD in this third phase, the MDs are again incompletely formed (Kobayashi et al, 2005). This third phase is also dependent upon the Pax2 gene; mice mutants for this gene show cellular invagination but no elongation because the WDs have degenerated (Torres et al, 1995; Miyamoto et al, 1997). Studies have also shown that the WD not only acts as a physical guide but also plays a role in MD elongation through paracrine signaling. More specifically, Wnt9b is expressed by the WD epithelium, and gene inactivation results in incomplete formation of the MDs (Carroll et al, 2005). Loss of Wnt9b expression did not affect the WD, per se, or the first two phases of MD development but did affect the caudal extension and elongation of the MDs, suggesting that WD signaling by Wnt9b is one of the critical factors in directing MD formation (Carroll et al, 2005). For a clinical correlation, see Anomalies in the Female (Unilateral Renal Agenesis section). (From Masse J, Watrin T, Laurent A, et al. The developing female genital tract: from genetics to epigenetics. Int J Dev Biol 2009;53:411–24.) Several genes play a critical role in WD development, including Pax2/8, Gata3, and Lim1. Many of the same genes affecting renal development will also affect internal duct development. If there is gene inactivation of Pax2/8, Gata3, or Lim1, there will be no formation of the kidneys (BRA), ureters, or genital tract (Uetani and Bouchard, 2009). The pathway for mammalian kidney development is regulated by reciprocal epithelial-mesenchymal inductive signaling between the UB epithelium and the MM (Yu et al, 2004). Ureteral bud formation and the induction of its branching require glial cell line–derived neurotrophic factor (GDNF), a secreted growth factor expressed in the MM (Michos et al, 2007). The GDNF ligand activates the RET receptor, which is expressed in the WD epithelium and then around the UB tips as branching proceeds. Gdnf expression and localization are positively regulated by Eya1 and Pax2 (Michos, 2009). GDNF activation of RET requires the glial cell line–derived neurotrophic factor family receptor α1 (GFRα1) and is essential for induction of UB formation and initiation of outgrowth and branching (Chi, 2009). Most genes that are thought to be essential for UB formation are also regulators of Gdnf or Ret expression. Studies of murine kidney development show that Gdnf −/− mice have renal agenesis, while Ret−/− mice have renal agenesis or dysplastic kidneys (Pichel et al, 1996; Schuchardi et al, 1996, Glassberg, 2002). Skinner and colleagues (2008) examined the association between abnormal kidney development and mutations of RET, GDNF, and GFRα1 in 29 stillborn fetuses with BRA or unilateral renal agenesis (URA). Mutations in RET were found in 7 of 19 fetuses with BRA and 2 of 10 fetuses with URA. A mutation in GDNF was found in 1 fetus with URA who also had mutations in RET. No GFRα1 mutations were observed. These data suggest that congenital renal agenesis results from RET mutations that prevent or impede the embryonic development of RET-dependent structures. After the GDNF/GFRα1 complex binds to RET, the Wnt11 gene is activated in the epithelial tips of the UB and is associated with UB branching (Majumdar et al, 2003). Wnt11 is a member of the Wnt gene family, which is composed of structurally related genes encoding secreted signaling proteins. These proteins are likely involved in several processes, including regulation of cell fate and patterning during embryogenesis. WNT11 signaling is required for the propagation of mesenchymal GDNF signaling, which establishes the autoregulatory epithelial-mesenchymal GDNF/WNT11 feedback signaling loop that controls the progression of metanephric branching morphogenesis after initiation of UB outgrowth (Majumdar et al, 2003; Michos et al, 2007). Bone morphogenetic protein-4 (BMP4), a member of the transforming growth factor-α family, is expressed in the periureteral mesenchyme and is essential for morphogenesis (Glassberg, 2002; Miyazaki et al, 2000, 2003; Michos et al, 2007). In wild-type mouse embryos, BMP4 is expressed by the mesenchyme surrounding the WD and UB. Mesenchymal cells that express BMP4 inhibit UB formation, in part by inhibiting Wnt11 expression. BMP4 migrates to ectopic sites, thereby preventing ectopic UB formation. The BMP4 mesenchymal cells act in a similar fashion during UB branching by inhibiting side branching and permitting stems to lengthen. Mesenchymal cells that are devoid of BMP4 surround the tip, where further branching proceeds. Mice heterozygous for a null mutation in BMP4 (+/−) manifest anomalies, including hypoplastic/dysplastic kidney, hydroureter, ectopic ureter, ureteral duplication, megaureter, ureterovesical junction obstruction, and reflux (Miyazaki et al, 2000, 2003). Gremlin 1 (GREM1) is an extracellular BMP antagonist that is expressed in the meso- and metanephric mesenchyme (Michos et al, 2004) (Fig. 117–2). GREM1 is upregulated in the mesenchyme around the origin of the UB prior to initiation of its outgrowth. BMP activity, at this time, is reduced locally. In the Grem1-deficient mouse embryo, metanephric development is disrupted at the stage of UB outgrowth initiation, resulting in bilateral renal agenesis. This inhibition of UB outgrowth causes progressive loss of Gdnf expression, resulting in apoptosis of the MM. (From Michos O, Goncalves A, Lopez-Rios J, et al. Reduction of BMP4 activity by gremlin 1 enables ureteric bud outgrowth and GDNF/WNT11 feedback signaling during kidney branching morphogenesis. Development 2007;134:2397–405.) To further examine the relationship of these various ligands and their effects on epithelial-mesenchymal interactions, Michos and colleagues (2007) cultured early Grem1-deficient mutant mouse kidney rudiments in medium supplemented with recombinant GREM1. The addition of GREM1 restored UB outgrowth and induced supernumerary epithelial buds that invaded the MM and initiated branching morphogenesis. At the molecular level, GREM1 replacement activated Wnt11 expression in the epithelial buds and upregulated Gdnf expression in the mesenchyme. Because genetic suppression of BMP4 activity in a Grem1-deficient mouse model completely restored kidney development, the local reduction of BMP4 activity by GREM1 was presumed to be critical to the initiation of UB outgrowth and kidney organogenesis. Because BMP4 signaling by the mesenchyme surrounding the WD prevents formation of supernumerary epithelial buds, successful initiation of UB outgrowth most likely requires both antagonism of BMP4 by GREM1 in mesenchyme and signaling by GDNF from the MM to RET in the ureteric epithelium (Michos et al, 2007). In addition, autoregulatory feedback signaling between GDNF in the mesenchyme and WNT11 in the epithelial tips regulates branching morphogenesis. Grem1 is essential for both upregulation of Wnt11 in the ureteric epithelium and Gdnf expression in the mesenchyme and the establishment of epithelial-mesenchymal feedback signaling. In an extensive autopsy analysis by Ashley and Mostofi (1960), the kidneys were completely absent on gross inspection of the entire retroperitoneum. Occasionally, there was a small mass of poorly organized mesenchymal tissue containing primitive glomerular elements with only minute vascular branches from the aorta. Complete absence of the renal vessels was observed in about 25% of specimens with BRA in this series. Complete ureteral atresia was observed in 39 of the 42 cases of BRA, and partial ureteral absence was noted in 3. Only small projections with no demonstrable lumen alongside the bladder were noted. With complete absence of the ureter, a rudimentary kidney was discovered in only a few instances, supporting the concept of reciprocal induction. The adrenal gland may appear flattened on ultrasonography but is rarely malpositioned or absent (Davidson and Ross, 1954; Hoffman et al, 1992). A normally located adrenal gland is expected, because the adrenal cortex develops from primitive mesoderm medial to the urogenital ridge and the medulla develops from ectodermal neural crest cells, while the metanephros is derived from the intermediate mesoderm. Fused and/or horseshoe-shaped glands have been noted on prenatal ultrasound screening (Strouse et al, 2002). Potter (1965) noted that fused glands were often found in the presence of spinal anomalies. In a small number of autopsies, the gonads were absent, indicating the abnormality or insult occurred before the fifth week and affected the overall development of the urogenital ridge (Carpentier and Potter, 1959). Key Points: Bilateral Renal Agenesis In the Ashley and Mostofi series (1960), about 50% of cases of complete ureteral atresia showed complete absence of the bladder, and in the remainder of cases, a hypoplastic bladder was found consisting only of a muscular tube with a minute lumen. In Potter’s series (1965), the bladders were also hypoplastic and lacked ureteral orifices. A normally closed urachus was observed. Abnormal development of the bladder is thought to be due to the lack of stimulation by fetal urine production, which starts at 10 to 12 weeks of gestation. Alternatively, it has been postulated that UB and WD structures migrating into the ventral cloacal region are needed to initiate normal bladder development. The absence of the UB, and not the lack of urine, may arrest bladder development (Levin, 1952; Katz and Chatten, 1974). This theory is supported by the fact that despite absence of bladder filling in bladder exstrophy, many of these bladders are functional following surgical closure alone, while the bladders associated with bilateral ureteral ectopia almost invariably require augmentation (Jayanthi et al, 1997, Gearhart and Matthews, 2007). Phenotypic features associated with BRA have been extensively described by Potter. These infants have low birth weights, ranging from 1000 to 2500 g, and intrauterine growth retardation due in part to low iron stores in the liver (Georgieff et al, 1996). At birth, oligohydramnios (absent or minimal amniotic fluid) is present. The characteristic facial appearance and deformity of the extremities distinguishes these children from normal newborns. The infants look prematurely senile and have “a prominent fold of skin that begins over each eye, swings down in a semi-circle over the inner canthus and extends onto the cheek” (Potter, 1946a, 1946b). This facial feature is a sine qua non of nonfunctioning renal parenchyma and suggests that its absence confirms the presence of at least one kidney (Fig. 117–3). The nose is blunted, and a prominent depression exists between the lower lip and chin. The ears appear to be low set, are drawn forward, and are often pressed against the side of the head, making the lobes seem unusually broad and exceedingly large. The ear canals are not dislocated, but the appearance of the ear lobes gives the impression that the ears are displaced downward. Periauricular pits and tags have been noted (Wang et al, 2001). The skin can be excessively dry and appears too loose for the body. This may be secondary to severe dehydration or lack of subcutaneous fat. The hands are relatively large and clawlike. The legs are often bowed and clubbed, with excessive flexion at the hip and knee joints (Saing et al, 1998; Carbillon et al, 2001; Das et al, 2002). Occasionally, the lower extremities are completely fused as seen with sirenomelia (Liatsikos et al, 1999). A lumbar meningocele with or without the Arnold-Chiari malformation and hydrocephalus is often observed (Davidson and Ross, 1954; Ashley and Mostofi, 1960). In Potter’s series (1965), anomalies of the gastrointestinal tract were found in 60% of fetuses. Anomalies of the external genitalia include absence of the scrotum and clitoral hypertrophy. Penile development is usually normal, but in a few cases, penile agenesis or a rudimentary penis and scrotum have been reported (O’Connor et al, 1993; Potter, 1965). Hypospadias is rare and does not appear to be related to the presence or absence of the testes. The testes are undescended in 43% of cases (Carpentier and Potter, 1959). Ashley and Mostofi (1960) found testicular agenesis in 10%. The vas deferens is normal in most cases, implying that the factor responsible for the renal agenesis influenced the UB only after it formed from a completely elongated WD or that the insult affected the induction of the MM. There is a relatively high incidence of anomalies of the MD structures and ovaries (Carpentier and Potter, 1959). The ovaries are frequently hypoplastic or absent. The uterus is usually rudimentary or bicornuate but occasionally absent. The vagina is a short, blind-ending pouch or completely absent. The characteristic facial abnormalities and limb features may result from deformations rather than malformations of structures due to the lack of “cushioning” from amniotic fluid (Fitch and Lachance, 1972; Thomas and Smith, 1974). This observation was confirmed by an experiment in nature in which one twin with BRA did not have the characteristic Potter facies because it shared the same amniotic sac with the second twin who had an adequate volume of amniotic fluid (Klinger et al, 1997). Fetal renal urine is the major source of amniotic fluid, accounting for more than 90% of its volume by the third trimester (Thomas and Smith, 1974; Chevalier and Roth, 2007), but the skin, gastrointestinal tract, and central nervous system also contribute small amounts, particularly before urine production begins at 10 to 12 weeks. Pulmonary hypoplasia and a bell-shaped chest are common associated findings that were originally thought to be due to uterine wall compression of the thoracic cage as a result of oligohydramnios (Bain and Scott, 1960). Subsequently, it was postulated that the amniotic fluid alone was responsible for pulmonary development (Fitch and Lachance, 1972). However, this theory was rejected when they observed a significant reduction in the number of airway generations as well as a decrease in acini formation in these fetuses (Hislop et al, 1979). Pulmonary airway branching occurs between the 12th and 16th weeks of gestation (Reid, 1977). A reduction in the number of branches implies interference with this process before the 16th week of gestation. Hislop and colleagues (1979) suggested that the anephric fetus fails to produce proline, which is a prerequisite for collagen formation in the bronchiolar tree. The kidney is the primary source of proline (Clemmons, 1977). Thus pulmonary hypoplasia may result from the absence of renal parenchyma and not from diminished amniotic fluid. This hypothesis is supported by the finding of normal lungs in two infants with prolonged leakage of amniotic fluid beginning at a time when pulmonary hypoplasia would have been expected if the amniotic fluid alone were responsible for the defect (Perlman et al, 1976; Cilento et al, 1994). Peters and colleagues (1991a) proposed a two-step process in pulmonary development, with a primary “renal growth factor” influencing early lung development and an amniotic fluid volume-dependent phase influencing later gestational lung growth. Smith and colleagues (2006) studied early lung development using a murine knockout model of renal agenesis/dysgenesis and anuria. They found that pulmonary development occurred early in embryogenesis, and fetal anuria and hypoplastic lung development preceded oligohydramnios. These observations support the two-step model proposed by Peters (1991a). Alternatively, oligohydramnios due to experimentally induced urinary obstruction is associated with pulmonary hypoplasia in fetal sheep, with initially normal renal function (Peters et al, 1991a, 1991b). Restoring amniotic fluid volume only partially restores lung growth. Therefore uropathy-associated pulmonary hypoplasia appears to be a result of oligohydramnios rather than renal dysfunction (Peters, 1991b). BRA is being diagnosed by prenatal ultrasonography in the second and third trimesters, when severe oligohydramnios is noted and no renal parenchyma can be identified (Forys et al, 2003). Termination of the pregnancy has been considered when the diagnosis is certain (Rayburn and Laferla, 1986). Additional diagnostic findings include small lung volumes and chest diameter and abnormal adrenal gland appearance (Latini et al, 1998; Sepulveda et al, 1998; Heling et al, 2001; Strouse et al, 2002). The characteristic Potter facies and the presence of oligohydramnios are pathognomonic. Amnion nodosum—small, white, keratinized nodules on the surface of the amniotic sac—have been considered a placental hallmark of prolonged, severe, oligohydramnios. Recently, oligohydramnios was diagnosed in only 22% of cases of amnion nodosum, suggesting that it may not be a reliable sign of oligohydramnios. Nevertheless, the finding portends a very poor prognosis (Adeniran and Stanek, 2007). Ninety percent of newborns void during the first day of life (Sherry and Kramer, 1955). In a study of 500 infants, every infant voided within the first 24 hours of life, regardless of the gestational age (Clarke, 1977). After the first 24 hours, anuria without distention of the bladder suggests BRA (Williams, 1974). However, most neonates with BRA who are born alive experience severe respiratory distress within the first 24 hours of life. When this becomes the focus of attention, the anuria may be initially unnoticed. Renal ultrasonography is the most efficient way to identify the kidneys and bladder and confirm the presence or absence of urine production. The advent of power Doppler ultrasonography has been highly accurate in determining the status of the renal arteries, even in fetuses with oligohydramnios and suspected BRA (Sepulveda et al, 1998). The finding of a flattened adrenal gland in its normal location supports the diagnosis of an absent kidney (Hoffman et al, 1992). If abdominal ultrasonography is inconclusive, renal scintigraphy can be performed using 99mTc-dimercaptosuccinic acid (DMSA). The absence of uptake of the radionuclide in both renal fossae above background activity or in an ectopic location confirms the diagnosis of BRA. Historically, umbilical artery catheterization and an aortogram were performed when other modalities were not diagnostic. About 40% of the affected neonates are stillborn. Of those neonates who are born alive, most do not survive beyond the first 24 to 48 hours due to respiratory distress associated with pulmonary hypoplasia. Survival subsequently depends on the rate at which renal failure develops. The longest-surviving child lived 39 days (Davidson and Ross, 1954). Complete absence of one kidney occurs more frequently than BRA but is not easily detected from findings on physical examination. An isolated single umbilical artery has been associated with renal anomalies, including unilateral renal agenesis (URA) (Dursun et al, 2005). More recently, the largest study to date of neonates with an isolated single umbilical artery did not find an increased incidence of URA or other malformations and concluded that postnatal renal ultrasonography was not routinely warranted (Deshpande et al, 2009). URA may remain undetected unless examination of the external genitalia and/or radiographic evaluation of the female or male pelvis for other reasons reveal an anomaly associated with renal agenesis. Over the past two decades, prenatal ultrasound examinations have been performed more routinely, and URA is being detected with increased frequency (Sipek et al, 1997). These imaging studies have also revealed that a substantial number of cases thought to be URA were a dysplastic or multicystic dysplastic kidney (MCDK) that had undergone involution prior to birth (Mesrobian et al, 1993; Hitchcock and Burge, 1994; Dell’Acqua et al, 2002; Hiraoka et al, 2002). A plain film of the abdomen supports this diagnosis if the splenic or hepatic flexure of the bowel is in its normal location and not in the ipsilateral renal fossa, suggesting that a dysplastic kidney or MCDK may have formed in the renal fossa before involuting. Curvilinear calcifications on a plain radiograph or computed tomography (CT) scan are another sign of a prior MCDK (Nakano et al, 1996). A flattened adrenal or the spleen (on the left) may be mistaken for a kidney in the 20-week structural ultrasound study, but at later gestational weeks, the diagnosis of URA becomes more apparent (Woolf and Hillman, 2006). Most autopsy series suggest that unilateral renal agenesis occurs once in 1100 births (Doroshow and Abeshouse, 1961). In an historical survey of excretory urograms, the incidence ranged between 1 in 1500 (Longo and Thompson, 1952) to 1 in 5000 (Wilson and Baird, 1985). Ultrasound screening of 280,000 school children in Taipei revealed the incidence of URA to be 1 in 1200 (Shieh et al, 1990). A similar incidence was found on prenatal screening in the Czech Republic (Sipek et al, 1997). The high male predominance of BRA is not nearly as striking in the unilateral condition, with a male to female ratio of 1.8 : 1 (Doroshow and Abeshouse, 1961). Absence of a kidney occurs somewhat more frequently on the left side. A familial tendency has been noted (Arfeen et al, 1993; Selig et al, 1993; Cascio et al, 1999). Siblings within a single family and even monozygotic twins have been affected (Kohn and Borns, 1973; Uchida et al, 1990). In a study of several families, McPherson and colleagues (1987) concluded the inheritance of URA was autosomal dominant with a 50% to 90% penetrance. This inheritance pattern has been confirmed by others who evaluated families with more than one affected individual (Biedel et al, 1984; Roodhooft et al, 1984; Battin et al, 1993). For screening recommendations, see the section Incidence under Bilateral Renal Agenesis. An absent kidney has been noted in a number of genetic disorders in which there is a deletion of several chromosomal loci: 8q13.3 (Pierides et al, 2002), 18q22.2 (Dowton et al, 1997), 22q11 (Anonymous, 1998; Stewart et al, 1999), as well as in X-linked and sporadic cases of Kallmann syndrome (Colquhoun-Kerr et al, 1999; Zenteno et al, 1999; Quinton et al, 2001). Several syndromes have been associated with URA, including Turner syndrome, Poland syndrome (Mace et al, 1972), Frazer syndrome (Fryns et al, 1997), BOR (brachio-oto-renal) syndrome (Pierides et al, 2002), DiGeorge anomaly (when associated with insulin-dependent diabetes mellitus in the mother) (Wilson et al, 1993; Novak and Robinson, 1994), dysmorphogenesis, and Kallmann syndrome. Abnormalities of the KAL1 locus at Xp22 in the X-linked autosomal-dominant disorder have a 40% incidence of URA (Say and Gerald, 1968; Colquhoun-Kerr et al, 1999; Zenteno et al, 1999; Quinton et al, 2001). Similarly, Townes-Brock syndrome with SALL1 deletions is associated with a high incidence of URA (Salerno et al, 2000; Nishinakamura et al, 2001; Sato et al, 2003, 2004). Twenty to 30 percent of children with the VACTERL association (Vertebral, imperforate Anus, Cardiac, Tracheo-Esophageal atresia, Renal, and Limb anomalies) have URA (Barry and Auldist, 1974; Kolon et al, 2000). Children with supernumerary nipples (Urbani and Betti, 1996) and disorders of the ears with hearing loss, especially if it is congenital (Huang et al, 2001), and preauricular pits (Pierides et al, 2002) have been thought to have an increased incidence of URA. Recently, studies have not shown a significant relationship between preauricular pits, minor ear tags, and URA (Arora and Pryce, 2004; Deshpande and Watson, 2006). Nonetheless, a screening renal ultrasonogram is recommended when these ear anomalies are found in the presence of other malformations. In addition, when more than one anomaly is present (e.g., ventricular septal defect [VSD] and an undescended testis) a screening renal ultrasonogram is prudent, but when specific complexes of anomalies associated with renal agenesis are present (for example, VACTERL-associated anomalies), a comprehensive radiographic review of all organ systems is mandatory. The embryologic basis for URA and BRA are thought to be similar. The etiology is most likely due to the UB, because increased RET mutations occur in humans with renal agenesis (Skinner et al, 2008). Complete absence of a bud or aborted ureteral development prevents reciprocal induction, which is critical for the development of the metanephric blastema into the definitive adult kidney. The metanephros is likely not to be responsible for the majority of cases, because the ipsilateral gonad (derived from adjacent mesenchymal tissue) is rarely absent, malpositioned, or nonfunctioning (Ashley and Mostofi, 1960). The high incidence of absent or malformed proximal WD structures in the male and anomalies of the MD structures in the female suggest that the embryologic insult affects the UB primarily in its early development and influences the development of WD derivatives. The abnormality most likely occurs no later than the fourth or fifth week of gestation, when the UB forms and the WD begins to develop into the ejaculatory duct, seminal vesicle, and vas deferens. The MD in the female begins its medial migration at this time, crossing over the WD (sixth week) during its differentiation into the fallopian tube, uterine horn and body, and proximal vagina (Woolf and Allen, 1953; Semmens, 1962; Yoder and Pfister, 1976). Magee and colleagues (1979) proposed an embryologic classification to explain the association of URA and MD anomalies (Fig. 117–4). In type I URA, the insult occurs before the fourth week, and there is nondifferentiation of the urogenital ridge structures, including the MD and WD. If unilateral, a uterus consisting of a single MD (unicornate uterus) will form and will be associated with contralateral renal agenesis. In type II URA, the insult occurs early in the fourth week of gestation, affecting both the WD and the UB. Because it is critical that the MD maintains close contact with the WD for MD elongation and subsequent fusion, maldevelopment of the WD affects renal development, MD elongation, contact with the urogenital sinus (UGS), and subsequent fusion. Therefore a didelphys uterus will form with obstruction of the horn and vagina on the side of the URA. Finally, in type III URA, the insult occurs after the fourth week, and the WD and MD elongation and differentiation proceed normally. In this case, only the UB and metanephric blastema are affected, thereby resulting in isolated URA. Figure 117–4 A to C, A proposed categorization of genital and renal anomalies in females. See text for details. (From Magee MC, Lucey DT, Fried FA. A new embryologic classification for urogynecologic malformations: the syndromes of mesonephric duct induced müllerian deformities. J Urol 1979;121:265.) The ipsilateral ureter is completely absent in about 60% of the cases (Fortune, 1927; Collins, 1932; Ashley and Mostofi, 1960). In the Ashley and Mostofi series, 19 of 232 with URA had only a portion of the lower end of the ureter present. There were no normally developed ureters reaching the level of the normal kidney. In most cases of complete absence of the ureters, the bladder showed no evidence of a ureteric orifice with failure of ipsilateral trigone development (Ashley and Mostofi, 1960). Cell lineage studies using a murine model show that the trigone has a urogenital sinus origin and should form normally (Viana et al, 2007; Mendelsohn, 2009). The trigone may not be distinguishable from the surrounding detrusor when the intramural ureter is absent. Therefore the endoscopic appearance of the trigone in this setting has lead to the probable misnomer in the case of the “hemitrigone” (in association with complete ureteral agenesis) or “asymmetrical trigone” (in the presence of a partially developed ureter). Segmental ureteral atresia on one side has been associated with contralateral ureteral or renal ectopia (Limkakeng and Retik, 1972). Except for ectopia or malrotation, anomalies of the contralateral kidney are infrequent (Longo and Thompson, 1952; Chow et al, 2005) (Fig. 117–5). However, abnormalities of the contralateral ureter are not uncommon, including ureteropelvic and ureterovesical junction obstruction in 11% and 7%, respectively (Cascio et al, 1999), and reflux in 30% (Atiyeh et al, 1993; Cascio et al, 1999). Other urologic abnormalities are found in 65% with URA (Kaneyama et al, 2004). Although the ipsilateral adrenal gland may be flattened (Hoffman et al, 1992), adrenal agenesis occurs in fewer than 10% of autopsy reports (Fortune, 1927; Collins, 1932; Ashley and Mostofi, 1960) and in 17% of individuals with URA undergoing a CT scan (Kenney et al, 1985). Genital anomalies are much more frequently observed. The incidence of a reproductive tract malformation for both sexes varies from 20% to 40% (Smith and Orkin, 1945; Doroshow and Abeshouse, 1961; Thompson and Lynn, 1966). Despite the predominance of males with URA, reproductive tract abnormalities in females occur in at least 25% to 50% compared with 10% to 15% in males. Regardless of sex, both gonads are usually normal. Therefore the different phenotypes that occur with URA may result from a primary urogenital ridge defect, which explains the finding of gonadal and adrenal agenesis in the minority of cases, or a primary defect in development of the UB and WD, which leads to the more common cases of URA and frequently observed abnormalities of the WD, MD, and their derivatives. The testis and head of the epididymis, which contain the efferent ductules derived from the mesonephric tubules, are invariably present; all structures proximal to that point, which develop from the WD (the body and tail of the epididymis, vas deferens, seminal vesicle, ampulla, and ejaculatory duct), are absent in almost 50% (Radasch, 1908; Collins, 1932; Charny and Gillenwater, 1965; Ochsner et al, 1972). Donohue and Fauver (1989) reported 79% of adult males with an absence of the vas deferens have an absent ipsilateral kidney; left-sided lesions predominated with a ratio of 3.5 : 1. However, bilateral absence of the vas has been noted with URA (McCallum et al, 2001). Occasionally, the WD structures are rudimentary or ectopic rather than absent (Holt and Peterson, 1974). Ipsilateral cryptorchidism rarely occurs. In 1914, Zinner reported a seminal vesicle cyst in association with ipsilateral renal agenesis (Pereira et al, 2009). Seminal vesicle cysts secondary to obstruction of the ejaculatory duct are currently diagnosed with increasing frequency as pelvic ultrasound examinations are performed more often (Lopez-Garcia et al, 1998; Kaneyama et al, 2004). Six cases (5%) were noted among 119 boys who were found to have URA during ultrasound screening of schoolchildren (Shieh et al, 1990). A pelvic ultrasonogram or magnetic resonance imaging (MRI) in boys diagnosed with URA may demonstrate a seminal vesicle cyst (Van den Ouden et al, 1998, Seo et al, 2009) (see Fig. 117–5). In cases of seminal vesicle cysts and URA, the ureter may insert into the prostatic urethra or seminal vesicle. Cystic dysplasia of the rete testis, a rare benign condition, is often associated with ispsilateral renal anomalies, most commonly URA (Wojcik et al, 1997; Camassei et al, 2002). A variety of anomalies may result in the female from incomplete MD formation because of alterations in normal WD development. Approximately one fourth to one third of women with URA have an abnormality relating to WD development (Thompson and Lynn, 1966; Heinonen, 2004). Conversely, 43% of women with genital anomalies have URA (Semmens, 1962; Heinonen, 1997). The most common MD anomalies are a true unicornuate uterus with complete absence of the ipsilateral horn and fallopian tube or a bicornuate uterus with rudimentary development of the horn on the affected side (Candiani et al, 1997) (see Fig. 117–5). The fimbriated end of the fallopian tube, however, is usually fully formed and is analogous to the head of the epididymis in the male (Shumacker, 1938). Partial or complete midline fusion of the MD may result in a double (didelphys) or septate uterus with either a single or a duplicated cervix (Radasch, 1908; Fortune, 1927). Complete duplication or separation of the vagina, proximal vaginal atresia associated with a small introital dimple, and complete absence of the vagina have been reported (Woolf and Allen, 1953; D’Alberton et al, 1981). Obstruction of one side of a duplicated system is not uncommon, and unilateral hematocolpos or hydrocolpos associated with a pelvic mass and/or pain has been described in pubertal girls (Weiss and Dykhuizen, 1967; Vinstein and Franken, 1972; Gilliland and Dick, 1976; Wiersma et al, 1976; Yoder and Pfister, 1976). Smith and Laufer (2007) suggested the acronym OHVIRA to classify the syndrome of Obstructed Hemivagina and Ipsilateral Renal Anomaly. In rare instances, this anomalous condition has been mistaken for a large or infected Gartner duct cyst. Sometimes a true Gartner duct cyst has been found in a prepubertal girl in association with an ectopic ureter that is blind ending at its proximal end or one that is connected to a rudimentary kidney (Currarino, 1982). Six percent of girls with URA were found to have a Gartner cyst on mass screening of schoolchildren (Shieh et al, 1990). Infertility occurs in as many as 33% of affected women with renal agenesis and unicornuate uterus (Heinonen, 1997). When specific anomalies of the uterus, including congenital absence of the uterus, unicornuate uterus, and didelphic uterus, are found on ultrasonography or MRI, radiologic investigation of the urinary tract often demonstrates URA or other renal anomalies (Bryan et al, 1949; Phelan et al, 1953; Thompson and Lynn, 1966; Candiani et al, 1997; Heinonen, 1997, Govindarajan et al, 2008; Reichman and Laufer, 2010). Another important anomaly often associated with URA is the Mayer-Rokitansky-Kuster-Hauser syndrome (MRKH), which is a complex of malformations occurring in 1 in 5000 newborn females (Guerrier et al, 2006). This syndrome not only includes renal anomalies but also genital tract anomalies ranging from upper vaginal atresia to total müllerian agenesis in an otherwise phenotypically normal female with a normal 46, XX karyotype. There are two subtypes reported. Type I is the typical form characterized by the finding of only symmetrical muscular buds or müllerian remnants and normal fallopian tubes. Type II, which is the more common but considered the atypical form, is characterized by asymmetrical hypoplasia of one or two buds with or without dysplasia of the fallopian tubes. Most importantly, the atypical form is often associated with renal anomalies, primarily URA or ectopia of one or both kidneys and horseshoe kidney in about 40% to 60% (Guerrier et al, 2006). In addition, there can be cervico-thoracic anomalies, auditory defects, and digital anomalies. Duncan and colleagues (1979) reported on the most severe constellation of malformations and referred to this as the MURCS association or MÜllerian duct aphasia (96%), Renal aphasia or ectopic (86%), and Cardiothoracic Somite dysplasia (two to four anomalous vertebrae between C5-T1 (80%). Anomalies of other organ systems are found frequently in affected individuals. The more common sites involve the cardiovascular (30%), gastrointestinal (25%), and musculoskeletal (14%) systems (Emanuel et al, 1974) (Fig. 117–6). They include septal and valvular cardiac defects, imperforate anus and anal or esophageal strictures or atresia, and vertebral or phalangeal abnormalities (Jancu et al, 1976; Wheeler and Weaver, 2001; Rai et al, 2002). Dursun and colleagues (2005) found that 44% of individuals with a congenital solitary kidney, most of whom had URA, had various nonurologic anomalies, but they detected lower incidences of these problems (cardiovascular, 15%; gastrointestinal, 9%; neurologic, 3%; and hematologic, 6%) than previously reported by Emanuel. Chow and colleagues (2005) reported a similar incidence of 42%. Key Points: Unilateral Renal Agenesis
Anomalies of Number
Bilateral Renal Agenesis
Syndromic Associations
Renal Embryology
Relationship of the Wolffian Duct to Müllerian Duct Formation
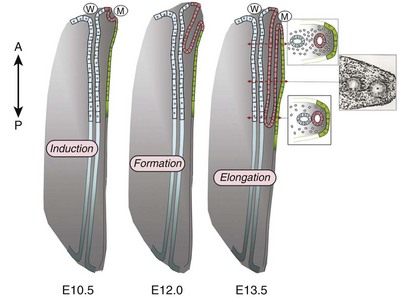
Molecular Mechanisms of Mammalian Kidney Organogenesis
Mammalian Kidney Organogenesis: New Advances
Gross Pathologic Description of Retroperitoneal Findings
Phenotypic Features
Role of Amniotic Fluid Production and Pulmonary Development
Prenatal and Postnatal Diagnosis
Postnatal Radiographic Evaluation
Prognosis
Unilateral Renal Agenesis
Incidence
Genetic/Syndromic Associations
Embryology
Associated Genitourinary and Adrenal Anomalies
Anomalies in the Male
Anomalies in the Female
Anomalies of Other Organ Systems
Diagnosis and Radiographic Evaluation
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Neuropathic Dysfunction of the Lower Urinary Tract
Neuropathic Dysfunction of the Lower Urinary Tract
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree