(1)
Division of Nephrology and Hypertension, Mayo Clinic, Rochester, MN, USA
(2)
Division of Hematology, Mayo Clinic, Rochester, MN, USA
Abstract
Amyloidosis is a group of diseases characterized by deposition of amyloid fibrils in soft tissues and organs. These fibrils all have the similar characteristics that distinguish them as amyloid. More than 24 proteins have been identified to form amyloid via several mechanisms such as genetic mutation, accumulation, age associated, and multifactorial. The kidney is one of the most commonly affected organs, but affinity to different organs may be predetermined by the native protein. The diagnosis of amyloidosis required demonstration of amyloid deposits in the tissues. In kidney tissue, amyloid appears as pale amorphous extracellular deposits that are periodic acid Schiff (PAS) and silver negative. Congo red staining will produce a green birefringence when viewed by polarized light. Amyloid fibrils are characteristically 7–12 nm in diameter and are randomly arranged on electron microscopy. Once amyloid is detected, typing must be performed to identify the native protein as treatment is type specific. The most common method of amyloid typing is with immunofluorescence or immunohistochemistry. If antibodies are not available, genetic testing may be helpful in identifying hereditary cases. The most advanced method of amyloid typing is liquid chromatography–tandem mass spectrometry. Once amyloid is identified, serum amyloid P (SAP) scintigraphy can be used to locate amyloid in the body. Treatment of amyloidosis depends on the type. Anti-myeloma therapy is used to treat immunoglobulin light chain amyloidosis (AL), antimicrobials or anti-inflammatory medications are used for serum amyloid A (AA), and organ transplantation may be used for some hereditary amyloidoses.
Introduction
Amyloidosis represents a group of diseases characterized by the deposition of amyloid fibrils in various tissues [1]. The result is progressive organ failure that can lead to death. These fibrils are 7–12 nm in diameter and are randomly arranged. They have the ability to take up Congo red dye and give off an apple-green birefringence when viewed with polarized light. This is a pathognomonic feature which separates amyloid from other fibrils which cause kidney disease.
To date, over 24 types of protein are known to cause amyloidosis in human [2]. The fibrils form as a result of misfolding which causes the protein to take up a pathologic confirmation that allows for subsequent self-aggregation. Several amyloidogenic mechanisms have been identified [1, 2]. In some, there is a natural propensity in the native protein to misfold. With these proteins, amyloid is formed as a result of accumulation or overproduction. Examples of this include senile amyloidosis (wild type transthyretin) which accumulates as a result of aging, dialysis-related amyloidosis (Aβ[beta]2m) where β[beta]2-microglobulin accumulates as a result of renal failure, and secondary amyloidosis (AA) where serum amyloid A is overproduced as a result of persistent inflammation or infection. Others are the result of a genetic mutation that produces an amyloidogenic protein (Table 21.1). Examples of this are (mutant) transthyretin (ATTR), fibrinogen α[alpha]-chain (AFib), lysozyme (ALys), apolipoprotein AI, and AII (AApoAI and AApoAII). Proteins can also become amyloidogenic after proteolytic modification. This is seen in Alzheimer’s disease with β[beta]-amyloid precursor protein (APP) and immunoglobulin light chain (AL). Prion which causes transmissible spongiform encephalopathies such as Creutzfeldt–Jacob disease, Gerstmann–Sträussler–Scheinker syndrome, fatal familial insomnia, and kuru is also considered an amyloidogenic protein (APrP). In some, multiple mechanisms may be involved. The best example of this is AL amyloidosis in which the monoclonal light chain is overproduced by the plasma-cell clones. These light chains undergo a partial proteolytic digestion, and some have mutations which may enhance the amyloidogenic potential of the monoclonal light chain [1].
Table 21.1
Mechanisms of amyloidogenesis
Age associated | Accumulation/overproduction | Genetic/hereditary | Multifactorial | Infectious | Unknown |
|---|---|---|---|---|---|
Senile | B2m | Transthyretin | AL | Prion | LECT2 |
Prolactin | SAA | Fibrinogen | AH | Beta | |
Calcitonin | Insulin | Lysozyme | ALH | IAPP | |
Amylin | Apolipoprotein AI | Calcitonin | |||
ANF | Apolipoprotein AII | Keratin | |||
Gelsolin | Localized AL | ||||
Cystatin C | |||||
BRI protein | |||||
Lactoferrin | |||||
SAA (HPFS) |
Different forms of amyloidosis have different organ tropism [2]. Some are quite specific such as the case of cystatin c (ACys) which causes the Icelandic form hereditary cerebral amyloid angiopathy resulting in cerebral hemorrhage, stroke, and dementia. Similarly, Aβ[beta] protein precursor (Aβ[beta]) is responsible for Alzheimer’s disease and is localized to the central nervous system. In others, skin is a preferential organ of deposition. Lichenoid or macular amyloidosis is the result of keratin (AD) deposition possibly as a result of trauma. AIns (insulin) can be found in diabetic patients near their insulin injection site. Nodular localized cutaneous amyloidosis is a form of localized AL that is often produced by polyclonal plasma cells probably in response to a localized inflammatory reaction. Systemic amyloidosis occurs when visceral organs are involved. These forms are often fatal as the persistent deposition of amyloid results in progressive organ failure (Table 21.2).
Table 21.2
Distribution by amyloid type
Systemic | Localized | Central nervous system |
|---|---|---|
AL | AL | Aβ[beta] |
AH | ACal | ACys |
AA | AIAPP | ABri |
AFib | AIns | ADan |
AApoAI | ALac (ocular) | APrpsc |
AApoAII | AD (pituitary) | |
ALys | APro | |
AGel | IAA/AANF | |
AB2m | ||
ATTR | ||
Senile |
Renal involvement is common in many forms of amyloidosis. Proteinuria frequently occurs and can be massive (>10 g/day) [3]. Renal insufficiency is variable and often depends on the severity of disease. Renal insufficiency with little proteinuria has also been described in a small subset of patients in which the amyloid preferentially deposits in walls of blood vessels instead of glomerular basement membranes or interstitium [4, 5]. Types of amyloid that involve the kidney include AL, AH (immunoglobulin heavy chain), AA, AFib, AApoAII, and ALECT2 (leukocyte cell-derived chemotaxin-2) [2, 6]. Renal involvement is uncommon in patients with ATTR which usually present with cardiomyopathy and neuropathy, but rare reports of renal involvement including end-stage renal disease have been reported. Finally, Aβ[beta]2m occurs only in dialysis-dependent patients due to a lack of clearance by dialysis [7]. Its main manifestations are soft tissue deposition (carpal tunnel) and arthropathy. However, autopsy studies have discovered systemic deposition usually in vascular beds including those of the kidneys. However, since these patients already had end-stage renal disease, the renal amyloidosis is never manifested.
Pathogenesis
The precursor amyloid proteins all share a common characteristic to misfold into β[beta]-sheet fibrillar protein [1]. This propensity can be natural or as a result of partial digestion as in the case of AL. The initial step involve formation of β[beta]-strands which allow the protein to be stacked on one another utilizing hydrogen bonds of alternating C=O and N–H groups. This self-assembly process eventually forms a sheet of polypeptides. The sheets are then mated together via a dry interface through a process known as the steric zipper [8]. In this cross-β[beta] model, sheets are mated together to form protofilaments. Four to six protofilaments are then braided together to form a fibril. Congo red dye is capable of intercalating the space formed between the protofilaments in a fibril which allows it to be used as a diagnostic tool. Ultrastructurally, fibrils from different precursor proteins are indistinguishable from one another despite the fact that precursor proteins come in different sizes and tertiary structures.
The pathogenic process of amyloid fibril is still not completely understood. Obviously, deposition of the amyloid fibrils certainly plays a big role. In cardiac amyloidosis, the heart is concentrically thickened resulting initially in diastolic dysfunction and later as disease progresses, systolic dysfunction. In the kidney, degree of proteinuria is associated with location of amyloid deposition while, glomerular filtration rate is determined by amount of deposition in the glomerulus. However, recent evidence suggests fibril deposition may not be necessary for cellular toxicity. Exposure to the precursor protein alone is sufficient for cytotoxicity to occur in cardiac myocytes [9]. The same phenomenon has also been noted in the kidney. Patients with massive proteinuria have been known to have very little amyloid deposits in their kidney. In fact, some of these patients were initially diagnosed as minimal-change disease [10]. Correct diagnosis was made when their biopsy was reviewed for failure to respond to steroids. Conversely, large amyloid deposits have been found in patients who responded to therapy and have normal proteinuria [11]. Electron microscopy of these patients suggests repair of the glomerular basement membrane can occur despite the presence of amyloid fibril [12].
Diagnosis
The diagnosis of amyloidosis requires the demonstration of the amyloid fibrils in the tissue (Fig. 21.1). The most commonly used method is Congo red staining [1, 3]. Congo red intercalates the fibrils and gives off an apple-green birefringence when viewed with polarized light. It is fairly sensitive but highly specific for amyloid fibrils. Thioflavin T is another stain that binds the β[beta]-sheet and gives off an enhanced fluorescence. However, it is considered to be less sensitive and specific than Congo red. Fibrils are also detected by electron microscopy. Characteristically, it is randomly arranged and has a diameter of 7–12 nm. These characteristics can be used to distinguish AL from other renal diseases with fibrillary deposits [13]. They include fibrillary glomerulonephritis, immunotactoid glomerulonephritis, cryoglobulinemia, hereditary nephropathies with fibronectin, and collagenofibrotic glomerulopathy. Fibrillary collagen can also be found in other glomerulopathies, most commonly in diabetes nephropathy, focal glomerulosclerosis, membranoproliferative glomerulonephritis, crescentic glomerulonephritis, and lupus. These diseases can be distinguished from amyloidosis by Congo red staining pattern and ultrastructural characteristics of the fibrils.
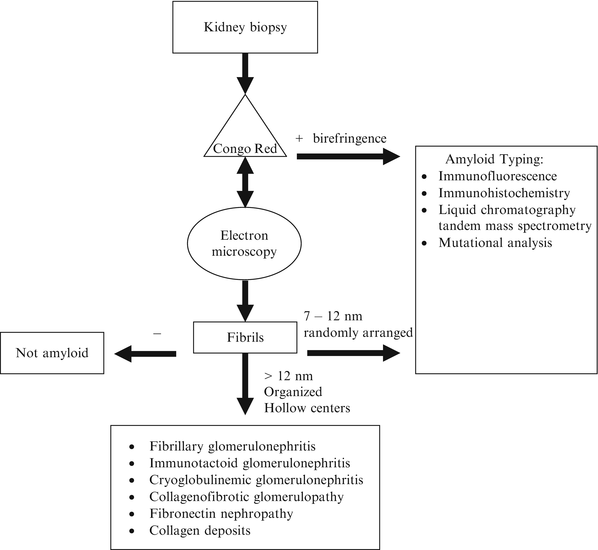
Fig. 21.1
Diagnostic approach for renal biopsies suspected of amyloidosis
Various tissues have been used for the diagnostic evaluation of amyloidosis. In patients with renal manifestations, amyloid can be detected on the kidney biopsy in virtually all cases [14]. Historically, renal biopsy was felt to be risky because of the possibility of amyloid angiopathy. Patients with AL can also develop an acquired factor X deficiency further increasing their risk of bleeding. However, a recent study of 101 patients with amyloidosis found the rate of post-biopsy hemorrhage was no different than patients without amyloidosis if they were approved by the standard screening tests used for all patients [15]. Other high-yield tissue included the heart, although it is much more invasive. Fat aspirate is often used. It is positive in approximately 70 % of the patients [14]. Rectal biopsy had been popular in the past. This is usually performed via a flexible sigmoidoscopy and has a sensitivity of 70–80 %. However, it is probably best to biopsy the organ which is symptomatic in order to maximize the odds of obtaining amyloid for typing.
In the kidney, amyloid can be seen in all three compartments. Amyloid initially appears along the glomerular basement membranes [3]. In more advanced cases, extensive deposition can be seen in the mesangium, becoming nodular in appearance in some cases. Vascular deposition is common, but in a small percentage of patients, it is the only place amyloid deposition occurs. On H&E stain, the deposits appear pink and amorphous. Amyloid deposits do not stain with periodic acid Schiff (PAS) or silver stain, but spicules can be seen along the glomerular basement membranes on silver stain (Fig. 21.2). In AL, immunofluorescence study should show a preferential staining for one of the light chains in areas where amyloid deposits have been identified. Light chain staining should be negative or equal and mild in intensity for all other types of amyloid.
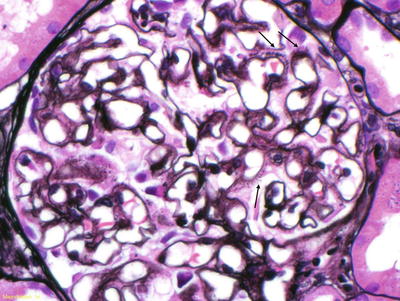
Fig. 21.2
Silver stain of a glomerulus. Arrows denote spicules along the glomerular basement membranes
Once amyloid is found in the tissue, typing of the amyloid protein is necessary. Treatment and prognosis differ for each subtype. Therefore, accurate typing is essential. Typing must be performed directly on the amyloid fibril. The use of surrogate markers such as circulating monoclonal protein or plasma-cell dyscrasia has led to misdiagnosis and treatment with cytotoxic agents [16]. It is paramount that AL or AH is confirmed before cytotoxic therapy is employed. Historically, potassium permanganate was used to distinguish AA from other forms of amyloid [3]. Applying potassium permanganate to the tissue prevents Congo red from binding to AA fibrils but not AL. However, immunohistochemical agents are now available. Antibodies to immunoglobulin light chains (κ[kappa] and λ[lamda]) and heavy chains, serum amyloid A protein, prealbumin (transthyretin), β[beta] 2-microglobulin, and fibrinogen are commercially available. Unfortunately, immunohistochemical identification is limited by the availability of the antibodies. Genetic testing has been used to identify many of the hereditary forms of amyloidosis. While this is helpful, caution is needed when interpreting the result. Differences in penetrance exist for different amyloidosis. The diagnosis is even more difficult when a monoclonal protein coexists or when dealing with senile amyloid where the amyloidogenic protein is wild type (non-mutated). Recently, the use of liquid chromatography–tandem mass spectrometry has made tremendous progress in the field of amyloid typing [17]. Tissues embedded on glass slides are dissected with a laser to capture amyloid-rich material. The tissue then undergoes tryptic digestion and is analyzed by the liquid chromatography–tandem mass spectrometry. The raw data are queried by multiple algorithms and the peptides are assigned a probability score. This technique allowed the identification of a new amyloid proteins [18]. The technique of SAP scintigraphy should be mentioned [19]. Serum amyloid P component is a molecule commonly found in all types of amyloid. Location and amount of amyloid deposits can be determined by injecting radiolabeled serum amyloid P component into the patient (Fig. 21.3). SAP scintigraphy is most useful in prognostication and assessment of response. And although a positive scan is virtually diagnostic for systemic amyloidosis, tissue biopsy is still required and should not be substituted [20].
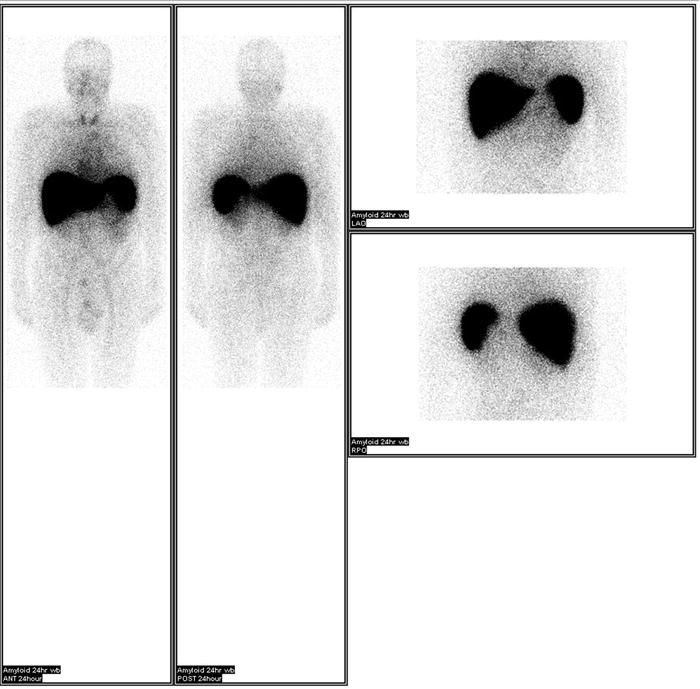
Fig. 21.3
SAP scintigraphy showing amyloid deposits in the liver and spleen. Images provided by Dr. Julian Gillmore
AL
Previously referred to as primary amyloidosis, AL is the most common form of amyloidosis in industrialized countries [14]. The age adjusted incidence ranges from 5.1 to 12.8/million/year which appears to be stable. The median age at presentation is 64 years with a range of 32–90 years. It is almost always the result of a plasma-cell dyscrasia, but rarely it can occur with a lymphoma or lymphoplasmacytic lymphoma (Waldenström’s macroglobulinemia). By immunofixation, a monoclonal protein is detectable in the serum and urine in 72 % and 73 % of patients, respectively. In the urine, the majority of the protein is made up of albumin with monoclonal protein representing only a small component. Eleven percent of the patients will have a negative monoclonal protein study by protein electrophoresis or immunofixation. These patients usually have a monoclonal light chain that exists in low levels. The sensitivity can be increased to 99 % by measuring serum-free light chain levels along with serum immunofixation [21]. Recent studies also showed serum-free light chain had a better correlation with outcomes of AL patients after treatment than the entire immunoglobulin [22]. Thus, measurement of serum-free light chain levels is essential in anyone suspected of having AL. A predilection toward lambda light chain exists in AL [14]. More than two-thirds of the patients with AL have a monoclonal lambda whereas the ratio is reversed in multiple myeloma. Approximately 18 % of the patients have >20 % plasma cells in the bone marrow, but coexistence with true multiple myeloma as defined by hypercalcemia, anemia, and lytic bone lesions is rare. AL is the most aggressive of the amyloidoses. Median survival is 13 months if left untreated with patients with advanced cardiac involvement or multiple myeloma faring the worst [23].
Kidney and heart are the most commonly affected visceral organs, but all visceral organs may be involved (liver, gut, spleen, and lung) as well as peripheral nerves, central nervous system, and soft tissues [14]. In a study of 474 patients, 73 % of patients presented with proteinuria and nearly half with renal insufficiency. The proteinuria was in the nephrotic range in approximately one-third of the patients. In a study of 145 patients, patients with lambda light chain were more likely to develop renal manifestations than ones with kappa [24]. The kappa to lambda ratio was 1:12 in patients with renal amyloidosis vs. 1:4 in those without (p = 0.02). Patients with lambda light chain also appeared to have more severe proteinuria. Median proteinuria of lambda patients was 3.6 g/day vs. 0.7 g/day in kappa patients (p = 001). When only patients with renal amyloidosis were analyzed, the disparity in proteinuria was maintained (7.2 g/day in lambda and 2.9 g/day in kappa). Elevated creatinine (p = 0.01) and higher proteinuria (p = 0.03) were risk factors for progression to end-stage renal disease (ESRD). ESRD eventually developed in 42 % of patients who presented with renal manifestations vs. 5 % of those without. This was similar to the rate (39 %) of ESRD reported in a recent Italian study of 198 biopsy-proven renal AL patients [25]. In the largest study to date with 923 patients from the UK, the rate of ESRD was slightly less at 23.9 % [26]. However, factors influencing progression to ESRD were similar (higher CKD stage and lower serum albumin). In this study, patients who had a hematologic response were less likely to require dialysis. Patients who progressed to ESRD had a significantly shorter survival [24]. Data for the USA and Europe showed a median survival of 11 months for patients with ESRD. These data may represent bias since approximately 20 % of the patients died within the first month many of whom withdrew from dialysis. A more recent study with more effective treatment found a median survival of 39 months with many patients surviving long enough to receive kidney transplantation [26].
Histologically, AL deposits can be found in all three compartments of the kidney [3]. Within the glomerulus, deposits may range from minimal to massive. Deposits can also be found in the interstitium as well as in the wall of blood vessels. Patients who have vascular limited deposits on renal biopsy have less proteinuria (<1 g) and typically present with unexplained renal insufficiency [4, 5]. The deposits in AL should stain preferentially for just one of the immunoglobulin light chains [3]. Immunofluorescence staining with antibodies to immunoglobulin light chains is therefore essential for the diagnostic evaluation. In AL variants, the deposits can be composed of immunoglobulin heavy chain (AH) or both immunoglobulin light and heavy chain amyloidosis (ALH) [27]. The immunoglobulin subclass should be identified to insure the heavy chain is monotypic. Antibodies are available for IgG, IgA, and IgM subclasses. In ALH, both the immunoglobulin heavy chain and light chain should be monotypic. Deposits in the kidney must match the isotype of circulating monoclonal protein detected in the blood or urine. In cases where the light chains are not well stained with immunofluorescence, immunoperoxidase may be helpful. Liquid chromatography–mass spectrometry (LC–MS) has been found to be very accurate in typing amyloidosis and is extremely helpful in cases where immunohistochemistry stains are unrevealing or equivocal. It is now considered the gold standard for amyloid typing (Fig. 21.4) [17].
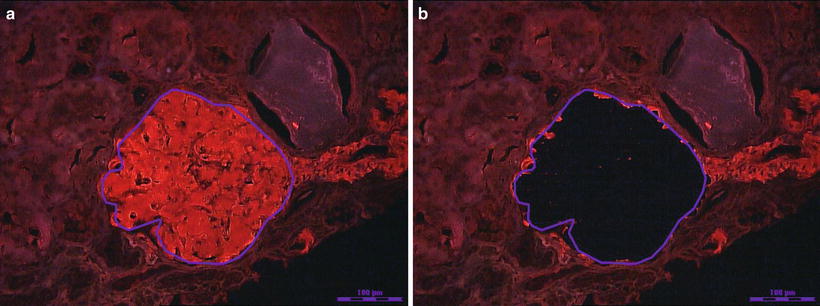
Fig. 21.4
(a) Photograph of a Congo red positive glomerulus traced for laser dissection and capture. (b) The glomerulus is dissected and captured for trypsin digestion and subsequent proteomic analysis by liquid chromatography and mass spectrometry. Photographs provided by Dr. Sanjeev Sethi
The treatment of AL had advanced considerably over the past two decades. The first effective treatment was melphalan and prednisone (MP). In two separate randomized controlled trials, MP extended median survival from 13 to 18 months [28–30]. Responders experienced significant reduction in proteinuria and alkaline phosphatase, but improvement in cardiac function was less common. Overall response rate was low in the 20–30 % range. Most of these patients achieved a hematologic partial response defined as >50 % reduction in the serum monoclonal protein, while hematologic complete response as defined by disappearance of the monoclonal protein was rare. In an attempt to reduce toxicity, high-dose dexamethasone was introduced. It was found to have some activity against AL. In a phase II trial, the median overall survival was 13.8 months [31]. The response was minor but was felt to be potentially beneficial if combined with other therapies. A median survival of 31 months was reported in patients treated with high-dose dexamethasone and undergone maintenance therapy with dexamethasone and alpha interferon [32].
The first therapy that significantly improved patient survival was high-dose melphalan followed by autologous stem cell transplantation (HDM–SCT) [33]. HDM–SCT was capable of achieving hematologic complete response at a high rate. In a study of 312 patients, a complete response was achieved in 40 % of the patients. In another large series of 270 patients, the partial response rate was 71 % with a complete response rate of 33 %. [34]. These high hematologic response rates helped extend the median overall survival to more than 4.6 years, and responders enjoy an even longer survival [33]. The factors associated with survival were the depth of the hematologic response and the severity of cardiac involvement at baseline. Improvement in proteinuria was noted in patients particularly those who had hematologic complete response. Of the patients with renal involvement, 63 % had a renal response after achieving a hematologic complete response. In the patients without complete response, the renal response rate of 11 %. Overall, 31.6 % of the patients with renal involvement had a renal response.
Despite its advantages HDM–SCT does have one major drawback, treatment-related mortality (TRM). This is defined as mortality within 100 days of initiating treatment. TRM can be as high as 40 % in some centers, but it is ~10 % at major amyloidosis centers [35, 36]. Patients with severe cardiac involvement, multiorgan involvement, or advanced age are at the highest risk. Unfortunately, organ involvement may be hard to define, and cardiac assessment based on echocardiography tends to be operator dependent. A risk assessment scoring system using cardiac biomarkers troponin T (cTnT) and N-terminal pro-brain natriuretic peptide (NT-pro-BNP) developed by the Mayo Clinic has been found to accurately predict TRM [37]. This significantly simplified the risk assessment since these biomarkers are easy to obtain and measure. They also would not be influenced by the operator. The use of this scoring system has been credited with lowering the TRM down to 8–9 % in some centers [38].
In early 2000, melphalan and dexamethasone (MDex) was found to be an effective treatment for AL. In a small study of 46 patients not eligible for HDM–SCT, 66 % achieved a hematologic response with 33 % complete response [39]. Long-term follow-up of these patients revealed a median overall survival of 5.1 years [40]. Renal response was noted in 48.3 % of the patients with renal involvement. Median time to hematologic response was 4.5 months. This is a concern since disease progression can occur before the therapeutic effects take place. To evaluate its efficacy against HDM–SCT, a randomized trial was conducted on 100 patients. This study found the median survival of MDex-treated patients (56.9 months) was longer than that of HDM–SCT-treated patients (22.2 months, p = 0.04) [41]. However, critics of the study pointed out that the HDM–SCT had extraordinary high early mortality. Ten patients died prior to HDM–SCT, and another 24 % of the 37 patients who underwent the procedure died within the first 100 days. Together 19 of the 50 patients randomized to HDM–SCT died within the first 130 days. Compare with the MDex group where seven patients died within the first 130 days with only two dying prior to therapy. The difference in mortality was most profound in patients considered high risk by the Mayo Clinic criteria (p < 0.0001). Overall survival was similar in patients considered low risk regardless of treatment (p = 0.12). The impact of the early mortality was demonstrated in the landmark analysis which found no differences in survival between the two groups after 6 months, p = 0.38. These results highlight the importance of patient selection when considering aggressive treatment in these patients. In one study, the TRM was reduced with 50 % with patient selection alone [42]. Thus HDM–SCT may still be useful in the treatment of AL, but its use should be limited to low-risk individuals [38].
Recently, two new classes of drugs have demonstrated efficacy against multiple myeloma and have made their way to AL therapy. The first are the immunomodulatory drugs (IMiDs) represented by thalidomide and lenalidomide. The other is bortezomib, a proteosome inhibitor. These novel agents have shown remarkable activity against multiple myeloma especially when used in combination with other agents [43]. Lenalidomide and dexamethasone produced an overall hematologic response rate of 67 % with a complete response rate of 29 % in a small study of 34 patients [44]. Forty-one percent of the patients with renal involvement had a renal response. Because lenalidomide is renally cleared, dosage adjustment was necessary. Significant treatment-related toxicities were noted with the usual dose of 25 mg a day. Most of the toxicities improved when the dose was reduced to 15 mg a day. Worsening of renal function was also noted in 59 % of the patients. Majority of these patients had renal involvement from AL. Serum creatinine increased to >2.0 mg/dL in 38 % of the patients. Thalidomide had been studied in combination with cyclophosphamide and dexamethasone. In this study, patients with New York Heart Association class II or higher or patients with significant fluid retention were started on a lower dose of thalidomide (50 mg a day vs. 100 mg a day) and dexamethasone (20 mg a day for 4 days vs. 40 mg a day for 4 days) [45]. Hematologic response rates were similar between the dosing regimens, and a hematologic response by free light chain criteria was achieved in 72 % of the patients with 32 % complete response. Overall survival after starting treatment was 41 months but had not been reached when calculated from diagnosis since majority of the patients had had prior treatments. Toxicity was significant. In the study, 40 % experience fatigue or somnolence and 21 % had worsening fluid retention or congestive heart failure. The combination of bortezomib and dexamethasone had been reported in small study with 18 patients from a single center [46]. Of the 16 evaluable patients, 94 % achieved a hematologic response with 44 % complete response. Renal response was noted in 14 % of patients with renal involvement. The major toxicities were thrombocytopenia, fatigue, neurotoxicity, and orthostatic hypotension. Two multicenter retrospective studies had been published. In one study of 26 patients, overall hematologic response rate was 54 % with complete response rate of 31 %. Organ improvement was noted in 12 %, stabilization in 76 %, and progression in 12 %. Thrombocytopenia and hyponatremia were reported as grades 3 and 4 toxicity, and grade 2 neurotoxicity was reported in 42 %. In the other with 94 patients, the hematologic response rate was 71 % with 25 % in complete response. Organ response was noted in 29 % of patients with heart involvement and 19 % of patients with kidney involvement. Hematologic response and reduction in NT-pro-GNP were independently associated with better survival [47]. In all of the reports involving therapies with novel agents, the follow-ups were relatively short. Longer follow-ups are needed in order to fully evaluate the true effectiveness of these therapies.
Related posts:
 General Approach to the Diagnosis and Management of Glomerular Diseases
Childhood Onset Nephrotic Syndrome
General Approach to the Diagnosis and Management of Glomerular Diseases
Childhood Onset Nephrotic Syndrome
 Membranoproliferative Glomerulonephritis
Membranoproliferative Glomerulonephritis
 Thrombotic Microangiopathies: Thrombus Formation Due to Common or Related Mechanisms?
Thrombotic Microangiopathies: Thrombus Formation Due to Common or Related Mechanisms?
 Glomerular Disease in Pregnancy
Glomerular Disease in Pregnancy
 Anti-glomerular Basement Membrane Disease
Anti-glomerular Basement Membrane Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


