TABLE 17.1 Genetic Classification of Alport Syndrome | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
AS cases. AR-AS results from mutations affecting both alleles of the COL4A3 gene or the COL4A4 gene. To date, most examples of AR-AS syndrome have resulted in early ESRD, but this may reflect ascertainment bias.15,16,17 Males and females are equally severely affected, and hearing and ocular defects are usual. It is not yet clear whether all AR-AS or all TBMN is caused by mutations of these genes.
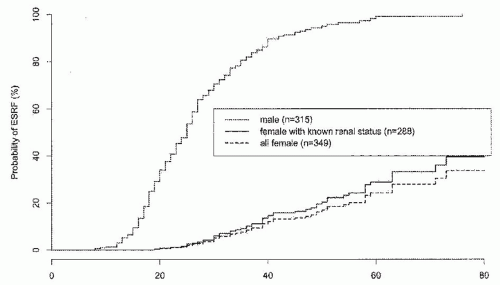 FIGURE 17.1 Probability of end-stage renal disease (ESRD) in 315 boys and men and 288 girls and women with the COL4A5 mutation. In the third curve, girls and women with incomplete clinical data were added because they were not in ESRD at last follow-up. (Reprinted from Jais JP, Knebelmann B, Giatras I, et al. J Am Soc Nephrol. 14:2603-2610;2003 with permission from the publisher, American Society of Nephrology.) |
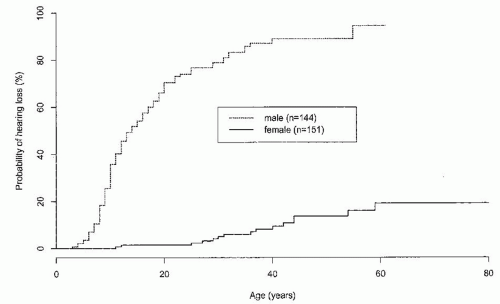 FIGURE 17.2 Probability of hearing loss in 144 boys and men and 151 girls and women with the COL4A5 mutation. (Reprinted from Jais JP, Knebelmann B, Giatras I, et al. J Am Soc Nephrol. 14:2603-2610;2003 with permission from the publisher, American Society of Nephrology.) |
is less resistant to degradation, thus resulting in the gradual deterioration of the GBM and in the clinical features of AS.31
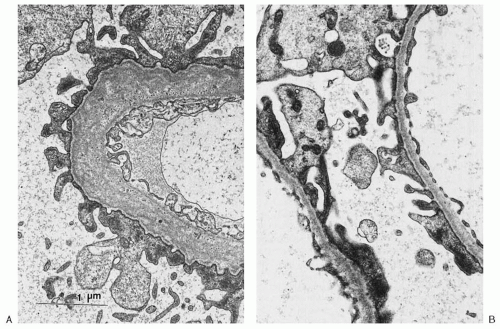 FIGURE 17.3 A: Glomerular ultrastructure in Alport syndrome. Electron micrograph of a renal biopsy specimen from a man in Utah kindred M, illustrating a widened lamina densa of the GBM (GBM). The lamina densa is split into several layers, between which may be seen numerous small electron-dense granules. B: Electron micrograph, at same magnification as (A), from an affected woman with familial thin GBM disease. The uniform thinning of the GBM can be appreciated by comparison with the width of the epithelial foot processes. (Electron micrographs courtesy of Dr. M.E. Hammond, University of Utah, Salt Lake City.) |
of GBM staining for α3(IV) and α5(IV) is indicative of AS: either XL-AS in male subjects or AR-AS in both genders.36 It should be noted that in 15% to 20% of XL-AS kindreds, α3(IV) and α5(IV) staining remains normal. This may result from the presence of certain missense mutations that cause only minimal disruption to the collagen chain structure. Both α3(IV) and α5(IV) are normally expressed in TBMN. Readers are referred to a comprehensive overview of differential α(IV) staining patterns for differentiation of the various forms of AS and TBMN by Haas.36
TABLE 17.2 Staining for α3(IV) and α5(IV) in Thin Basement Membrane Nephropathy (TBMN) and Alport Syndrome Variants | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
of heterozygous females.7 Hematuria is a sine qua non for the diagnosis of AS in males. Single or recurrent episodes of gross hematuria have been reported in 60% to 70% of males.4 They may follow sore throats or other infections in children and may be the presenting symptom.44
not practical for routine diagnosis due to the need to study several affected family members. About 15% of XL-AS cases are due to new mutations and, therefore, the family history will be negative.2,62 In these patients, AS is diagnosed or suspected based on a renal biopsy performed for unexplained hematuria with or without proteinuria.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


