Essentials of Diagnosis
General Considerations
Acute kidney injury (AKI) is a life-threatening disease process occurring in approximately 5% of all hospitalized patients and accounting for up to 30% of the admissions to intensive care units. AKI is preferred to acute renal failure as both kidney and injury are more patient-appropriate terms. Patients with AKI, regardless of their associated comorbid conditions, have a greater than 5-fold increased mortality rate. AKI is characterized by a reduction in the glomerular filtration rate (GFR) resulting in retention of nitrogenous wastes (creatinine, BUN, and other molecules that are not routinely measured). Early in the course of AKI patients are often asymptomatic and the condition is diagnosed only by observed elevations of BUN and serum creatinine levels or oliguria. An initial rise in serum creatinine of 0.5 mg/dL or a 25% increase in serum creatinine is often used to define AKI, although there is no definitive definition.
Oliguria (urine output less than 400 mL per 24 hours or 15 mL per hour) occurs commonly in AKI and may be an important indicator of renal dysfunction. However, urine output cannot be the only measure of kidney function. Patients with nonoliguric AKI usually have a better prognosis primarily due to less severe injury and/or a higher incidence of nephrotoxic-induced AKI in the nonoliguric group. Unfortunately, there has been little improvement in survival from AKI since the advent of hemodialysis and the mortality remains greater than 50% in many studies.
Definition
The RIFLE criteria consists of various graded levels of kidney injury based upon percent rise in serum creatinine, urine output and outcome measures.
Risk: 1.5-fold increase in the serum creatinine or GFR decrease by 25 percent or urine output <0.5 mL/kg per hour for six hours
Injury: Twofold increase in the serum creatinine or GFR decrease by 50 percent or urine output <0.5 mL/kg per hour for 12 hours
Failure: Threefold increase in the serum creatinine or GFR decrease by 75 percent or urine output of <0.5 mL/kg per hour for 24 hours, or anuria for 12 hours
Loss: Complete loss of kidney function (eg, need for renal replacement therapy) for more than four weeks
ESRD: Complete loss of kidney function (eg, need for renal replacement therapy) for more than three months
The AKIN (Acute Kidney Injury Network) criteria are a modification of the RIFLE criteria and include both diagnostic and staging system.
Stage 1. Increase in serum creatinine ≥0.3 mg/dl or 1.5 to 2 fold increase from baseline or urine output less than 0.5 mL/kg per hour for more than 6 hours
Stage 2. Increase in serum creatinine >2-3 folds from baseline or urine output less than 0.5 mL/kg per hour for more than 12 hours
Stage 3. Increase in serum creatinine >3 fold from baseline or serum creatinine of ≥4.0 mg/dl with an acute rise of at least 0.5 mg/dl or urine output less than 0.3 mL/kg per hour for 24 hours or anuria for 12 hours.
Pathogenesis
Prerenal azotemia, the most common cause of AKI, accounting for 30–50% of all cases, is characterized by a diminished renal blood flow, primarily due to decreased effective arterial blood flow (Table 9–1). By definition, prerenal azotemia is a rapidly reversible process if recognized early and the underlying cause of reduced renal blood flow is corrected. Prerenal azotemia occurs when there is a reduction in the effective arterial blood flow to the kidney, either from an absolute reduction in the volume of extracellular fluid (eg, hypovolemia) or in conditions in which the effective circulating volume is reduced despite a normal total extracellular fluid volume (eg, congestive heart failure). Effective arterial blood flow is the amount of arterial blood perfusing vital organs. The determinants of effective arterial blood flow include the actual arterial volume, cardiac output, and vascular resistance. It is important to realize that the extracellular fluid (ECF) volume and/or venous volume may have no relationship to effective arterial volume. Although venous and ECF volumes can be accessed by careful physical examination, effective arterial volume cannot. Therefore, in certain circumstances clinicians must rely on additional information beyond the physical examination to ascertain a measure of organ perfusion. Invasive cardiac monitoring and determination of the renal fractional excretion of Na+ (FeNa+) are the useful estimates of effective arterial circulatory volume.
Etiology | Mechanism | Extracellular fluid volume |
|---|---|---|
Hemorrhage | True intravascular volume depletion | Reduced |
Burns | ||
Diuretics | ||
Dehydration | ||
Gastrointestinal losses | ||
Vomiting | ||
Diarrhea | ||
Pancreatitis | ||
Nasogastric suctioning | ||
Enteric fistula | ||
Congestive heart failure | Decreased effective circulating volume | Increased |
Cardiac tamponade | ||
Aortic stenosis | ||
Cirrhosis with ascities | ||
Nephrotic syndrome | ||
Angiotensin-converting enzyme inhibitors | Impaired renal blood flow | Normal |
Nonsteroidal anti-inflammatory drugs | ||
Renal artery stenosis | ||
Renal vein thrombosis | ||
Sepsis | Systemic vasodilation | Normal |
Vasodilatory drugs | ||
Anesthetic agents |
The fractional excretion of sodium is calculated as follows:
An FeNa+ of less than 1%, in the setting of an increasing serum creatinine or BUN, is generally indicative of prerenal azotemia as the reduced, renal blood flow results in a sodium avid state. In patients with prerenal azotemia, proximal tubule cells are undamaged and continue to function appropriately to avidly reabsorb Na+ and water. Due to increased proximal reabsorption of Na+ there is decreased distal delivery of Na+ leading to increased renin secretion. This mediates enhanced aldosterone synthesis resulting in increased distal Na+ reabsorption. The end result is a low FeNa+ (<1%). Exceptions to this rule, resulting in a high FeNa+ with prerenal azotemia, include use of diuretics within the previous 24 hours, glucosuria, metabolic alkalosis with high urinary bicarbonate, obligatory loss of Na+, and chronic kidney disease with a high baseline Na+ excretion. A low FeNa+ is also seen in the early stages of acute glomerulonephritis, urinary obstruction, pigment nephropathy, and AKI induced by radiocontrast agents.
Low effective arterial volume states also stimulate release of antidiuretic hormone (ADH), leading to increased distal urea and water reabsorption. A low fractional excretion of urea nitrogen (<35%) can be especially useful in states of high urinary flow when prerenal azotemia occurs as in cases of high solute administration, such as in burn and trauma patients. The BUN to serum creatinine ratio, which is usually 10:1, also increases (>20:1) as filtered urea is reabsorbed and creatinine is excreted in prerenal azotemia.
The primary pharmacologic agents causing prerenal azotemia include angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), and nonsteroidal anti-inflammatory drugs (NSAIDs) including Cox-2 inhibitors. ACE inhibition results in a decreased GFR due to dilation of the efferent arteriole and reduction in glomerular filtration pressure. In certain patients (eg, those with bilateral renal artery stenosis) the GFR is particularly dependent on the effects of angiotensin II. If these patients take an ACE inhibitor, their GFR decreases even though renal blood flow is not reduced. NSAIDs cause prerenal azotemia by blocking the intrarenal vasodilatory effect of prostaglandins. They should be avoided in patients with reduced effective arterial volume including patients with congestive heart failure, liver disease, nephrotic syndrome, and preexisting renal dysfunction.
Intrinsic AKI is subdivided into four categories: tubular disease, glomerular disease, interstitial disease, and vascular disease (Table 9–2).
Etiology | Examples |
|---|---|
Tubular ischemia and inflammation (moderate to severe) | Shock, sepsis, bypass surgery |
Nephrotoxins | Aminoglycosides, cisplatin, heme pigments, radiocontrast agents, nonsteroidal anti-inflammatory drugs, cyclosporine A |
Small vessel vasculitis | Pauci-immune glomerulonephritis, hemolytic uremic syndrome |
Acute glomerular nephritis | Rapidly progressive glomerulonephritis, infective endocarditis |
Interstitial nephritis | Methicillin, any drug |
Tubular obstruction | Uric acid, methotrexate, acyclovir, sulfonamides |
Acute tubular cell injury is the most common cause of intrinsic AKI, accounting for approximately 90% of all hospital acquired AKI. Acute tubular necrosis (ATN) is the common term used for this type of AKI, which is usually induced by ischemia, sepsis, or toxins. Acute tubular dysfunction resulting from tubular cell injury is far more common than true cellular necrosis. ATN is usually reversible unless the ischemia was severe enough to cause cortical necrosis, which is associated with severe oliguria or anuria and is rare.
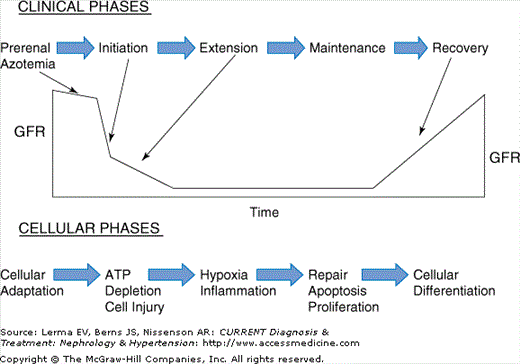
Tubular cell injury and death are important contributors to alterations in GFR following ischemic injury through several mechanisms. Figure 9–1 outlines the pathophysiology and clinical phases of ischemic AKI. In the initiation phase of AKI there is ATP depletion resulting in proximal tubule, endothelial, and smooth muscle injury and apoptosis. The extension phase of AKI occurs with persistent ischemia, vascular congestion, and ongoing hypoxia. Endothelial damage and activation result in an imbalance in vasoactive mediators and persistent vasoconstriction, particularly in the outer medulla. These mediators and endothelial damage lead to an increase in permeability, which increases interstitial pressure and decreases capillary blood flow. This results in continued hypoxia during reperfusion and enhanced tubular cell injury and death via apoptosis in this area. The end result of these various pathophysiologic processes is further worsening of the GFR. The extension phase is followed by a prolonged maintenance phase in which BUN and creatinine continue to rise. If there is no further injury the recovery phase begins in 1–2 weeks. Apoptosis occurs in all phases leading to remodeling of injured tubules and facilitating their return to a normal structural and functional state. Most cells recover by cellular repair. However, some epithelial cells dedifferentiate, replicate, and migrate to fill the epithelial defect. Thereafter they spread out, become attached to the tubular membrane, and reestablish their polarized differentiated structure.
AKI occurs in approximately 20-25% of patients with sepsis and 51% with septic shock. The combination of AKI and sepsis is associated with a 70% mortality, as compared with a 45% mortality among patients with AKI alone. Thus, the combination of sepsis and AKI constitutes a particularly serious medical problem. There is experimental evidence that early in sepsis-related AKI the predominant pathogenetic factor is renal vasoconstriction with intact tubular function, as demonstrated by increased reabsorption of tubular sodium and water. Thus, intervention at this early stage may prevent progression to AKI and cell injury. Renal vasoconstriction in sepsis seems to be due, at least in part, to the ability of tumor necrosis factor to release endothelin. Endothelial damage, endotoxemiageneration of oxygen radicals, complement pathway activation, and disseminated intravascular coagulation may all contribute to the pathophysiology of ischemic AKI.
Since the early vasoconstrictor phase of sepsis and AKI is potentially reversible, it should be an optimal time for intervention. However, clinical studies performed in patients up to 72 hours after admission to the intensive care unit, in which attempts were made to optimize hemodynamics and monitor the patients with a pulmonary artery catheter, not only were negative but showed increased mortality among patients with sepsis. In contrast, a randomized study of over 200 patients showed that early goal-directed therapy during the first 6 hours after admission was effective. In patients treated with this approach, the multiorgan dysfunction score and in-hospital mortality was decreased significantly compared with four patients who received standard care. The goal-directed approach included early volume expansion and administration of vasopressors to maintain mean blood pressure at or above 65 mm Hg and transfusion of red cells to increase the hematocrit to 30% or more if central venous oxygen saturation was less than 70%. If these interventions failed to increase central venous oxygen saturation to greater than 70%, therapy with dobutamine was instituted.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


