Medication-related AIN
AIN associated with infections
Direct invasion of the kidney
Remote infection
Idiopathic AIN—TINU syndrome
AIN associated with glomerular and systemic diseases
With anti-TBM antibodies
SLE and MCTD
Sarcoid
Sjogren’s
Close to 100 different medications have been incriminated in producing AIN, with antibiotics of the beta-lactam class (penicillins and cephalosporins) frequently cited [18–21] (see Table 23.2). In particular, in over 100 patients methicillin has been reported to cause AIN. AIN has also been reported with numerous other penicillins and cephalosporins including both second and third generation cephalosporins. Despite wider usage of other antimicrobials and discontinuation of the use of methicillin, cases of beta-lactam-induced AIN continue to be reported. A recent study documented four cases of nafcillin-induced AIN within 1 year in a single institution [22]. Other antimicrobials including the sulfonamides, rifampin, and the quinolones have all clearly produced AIN [1, 3–10]. Vancomycin has also been identified as an inciting agent in AIN [23, 24]. While the antiviral agents, acyclovir and foscarnet, can also produce tubulointerstitial damage, this manifestation is more similar to acute tubular necrosis than to true interstitial nephritis [3, 25–28].
Table 23.2
Medications associated with AIN
Beta-lactam antibiotics |
|---|
Methicillina |
Penicillin |
Ampicillin |
Apacillin |
Flucloxacillin |
Oxacillin |
Mezlocillin |
Nafcillin |
Carbenicillin |
Amoxicillin |
Mezlocillin |
Cefaclor |
Cefotaxime |
Cefoxitin |
Cefuroxime |
Cephalothin |
Cephalexin |
Cephradine |
Cephaloridine |
Other antimicrobials |
Sulfonamidesa |
Trimethoprim-sulfamethoxazolea |
Rifampina |
Polymyxin |
Tetracyclines |
Vancomycin |
Isoniazid |
Chloramphenicol |
Ethambutol |
Minocycline |
Erythromycin |
Para-aminosalicylic acid |
Ciprofloxacina |
Norfloxacin |
Piromidic acid |
Spiramycin |
Nonsteroidal anti-inflammatory agents |
Ketoprofena |
Fenoprofena |
Indomethacin |
Ibuprofen |
Benoxaprofen |
Phenazone |
Mefenamic acid |
Tolmetin |
Diflunisol |
Zomepirac |
Piroxicam |
Diclofenac |
Suprofen |
Oxyphenbutazone |
Novominsulsone |
Diuretics |
Thiazides |
Furosemide |
Ethacrynic acid |
Chlorthalidone |
Triamterene |
thiazide/amiloride |
Other medications |
Phenindionea |
Glafeninea |
Diphenylhydantoina |
Cimetidinea |
Sulfinpyrazonea |
Allopurinola |
Proton-pump inhibitorsa |
Carbamazepine |
Clofibrate |
Azathioprine |
Aspirin |
Phenylpropanolamine |
Aldomet |
Phenobarbital |
Leukocyte interferon A |
Haldol |
Coumadin |
Tofranil |
Diazepam |
Valproic Acid |
Chlorprothixene |
Captopril |
Propranolol |
Amphetamines |
Doxepin |
Quinine |
Ranitidine and cimetidinea |
In addition to antimicrobials, a number of different structural classes of diuretics including thiazides, the loop diuretics, and triamterene may infrequently produce AIN [1, 3–10]. All classes of nonsteroidal anti-inflammatory drugs including Cox-2 inhibitors have also been associated with AIN [29, 30]. There are also increasing reports of AIN related to a variety of proton-pump inhibitors [31]. Of the many other pharmaceutical agents producing AIN, the only frequently offending agents are allopurinol, diphenylhydantoin, sulfinpyrazone, cimetidine and ranitidine, and 5-aminosalicylates such as azulfidine and mesalamine [32–34].
While some infectious agents produce parenchymal damage by direct invasion of the kidney (e.g., acute bacterial pyelonephritis), others may be associated with inflammatory interstitial nephritis indirectly related to a systemic process. Such remote or systemic infections have been caused by bacteria, parasites, and viruses. Bacterial organisms associated with AIN include Streptococci, Brucella, Pseudomonas, Staphylococci, Legionella, Yersinia, Mycobacterium leprae, and Mycoplasma [1, 13–15, 35–41]. Leishmaniasitic parasitic infections including Kala-azar have also been associated with AIN as have cases of toxoplasmosis [42–44]. Epstein–Barr virus and measles infections have been associated with an interstitial renal inflammatory infiltrate [45–47]. One to two percent of patients with severe mononucleosis develop acute renal insufficiency which may be associated with biopsy documented AIN. Up to a third of biopsied patients with Kawasaki’s disease have an inflammatory reaction typical of AIN [48]. While in HIV infection with or without glomerular lesions, prominent tubulointerstitial damage is common, its presence without glomerular pathology should prompt a search for invading infectious agents or medication-related toxicity [3, 49].
For many other infectious agents associated with renal dysfunction, it is unclear to what extent the interstitial inflammation relates to direct tissue invasion as opposed to remote effects of the infectious organisms. This is true for reports of AIN associated with leptospirosis, mycobacterial infections, rickettsial infections, and viral infections due to herpes, CMV, hantavirus, polyomavirus, and adenovirus. In addition, an acute interstitial infiltrate is a common finding in both sarcoidosis and Sjogren’s syndrome [50–53].
Also AIN may be associated with a number of primary glomerulopathies [1, 3]. AIN with anti-TBM antibodies has occurred in a number of glomerulonephritides including Goodpasture’s syndrome, membranous nephropathy, and familial nephritis. AIN may be associated with immune complex deposits along the TBM in systemic lupus and mixed connective tissue disease [1]. In SLE interstitial damage may occur along with the glomerular involvement or more rarely as an isolated occurrence [1, 54, 55]. TINU (tubulointerstitial nephritis and uveitis) syndrome is a unique form of idiopathic AIN [56, 57]. Recently, a new entity of hypocomplementemic tubulointerstitial nephritis has been reported [58, 59].
Pathogenesis and Pathophysiology
Despite much speculation, the pathogenesis of medication-related AIN is complex and remains to be defined [1–10]. There are no good experimental models. It is unclear why AIN results more frequently with some medications of a given class (e.g., methicillin) than with others (e.g., penicillin) [1–10]. Neither the medication concentration in the serum of many patients with medication-related AIN protein binding, route of excretion by renal tubular secretion, nor the type of underlying infection seems an adequate explanation for the wide discrepancy in incidence. Moreover, it is unknown what factors predispose individuals to develop AIN in response to medications previously taken without causing problems. For example patients developing AIN associated with NSAID use typically have taken the medication for prolonged periods of time before developing the adverse renal reaction [60, 61]. In one series several patients had more than one episode of AIN implying some form of predisposition [13, 62].
In many patients with medication-related AIN, there is clear evidence for an immunologic reaction [1–10]. The adverse renal reaction is uncommon, not dose related, and suggestive of an allergic immune response. In those with hypersensitivity features such as rash, fever, eosinophilia, and eosinophiluria all suggest an allergic immune response. The pathology also supports an immunologic reaction by both the nature of the inflammatory cellular infiltrates and in some cases evidence of tubular basement membrane deposit (TBM) deposits. Prompt recurrence after rechallenge with the same or a similar drug also suggests an immune mechanism [62, 63]. A number of experimental models of tubulointerstitial nephritis unrelated to medications also suggest potential immunologic mechanisms of damage [1, 5].
The induction of AIN may relate to the kidney’s excretory role for many drugs, with binding of drug hapten to kidney tubular or interstitial structural proteins [1–10, 63]. Although the small molecular weight of many medications makes them capable of eliciting only a weak immunologic response, when bound as haptens to other proteins, they may elicit a stronger response. Indeed, in some cases of penicillin AIN, the benzylpenicilloyl hapten and, for some cases of methicillin AIN, the dimethoxyphenyl penicilloyl hapten have been localized along the TBM’s bound to kidney structural proteins [19, 20, 63–65]. However, these haptens have also been found bound to the interstitium of patients receiving these drugs, but who have not developed AIN [65]. It appears that under certain conditions in predisposed individuals, the drug-protein conjugate may initiate a response leading to AIN.
There is evidence for both humoral and cell-mediated immunologic reactions. Humoral mechanisms are supported in some patients by evidence of circulating anti-TBM antibodies [1, 59, 63, 65], while others have had circulating antibodies, which have bound to proximal tubule brush border antigens [66, 67]. In rare patients, immunofluorescence microscopy has confirmed the linear deposition of immunoglobulin or complement localized to the TBMs [19–21, 64, 68–71]. Rarely serum complement levels are depressed in medication-related AIN [18]. Increased serum IgE levels and interstitial inflammatory infiltrates with IgE-containing plasma cells are sometimes found, but despite these reported abnormalities, the majority of patients with medication-related AIN have no such findings [72–74].
A cell-mediated mechanism is supported by the prominent cellular interstitial infiltrates with lymphocytes and macrophages [1, 3]. A delayed hypersensitivity reaction to intradermal drug injection has also been occasionally documented [19, 64]. In NSAID-related AIN, the associated minimal change nephrotic syndrome has been cited as evidence of a cell-mediated lymphokine-directed reaction [27, 28, 60, 61]. The interstitial inflammatory infiltrates in medication-related AIN are composed predominantly of T lymphocytes rather than B lymphocytes, with a variable ratio of cytotoxic-suppressor to the helper-inducer T cell population [75–77]. There has been no difference in the individual T cell subtypes or T/B cell ratio between patients with beta-lactam-related AIN and those with NSAID-related AIN [77]. Drug-specific T cell clones may be involved in the pathogenesis of the lesions [78]. Although eosinophils are found in the biopsies of many patients with AIN, their role is also unclear [3]. They may be recruited by eosinophilic chemotactic factors and subsequently contribute to the interstitial damage through release of proteases, leukotrienes, superoxide radicals, and peroxidases [1–10].
The reduction in the glomerular filtration rate commonly seen probably relates directly to the inflammatory interstitial infiltration since the severity of the AIN correlates with the severity and diffuse nature of the inflammation. Interstitial edema may contribute to elevated intratubular pressures, and sloughed luminal cells and debris may lead to intratubular obstruction. The role of tubular back-leak across damaged epithelium, renal vasoconstriction, and tubuloglomerular feedback is unclear [72]. Structural damage from cellular infiltration and “tubulitis” or from release of mediators of inflammation may also play a role in the tubular defects seen in patients with medication-related AIN.
Pathology
The pathology of medication-related AIN is characterized by patchy to diffuse interstitial inflammatory infiltrates with variable amounts of edema and focal tubular damage [1–10, 13, 18, 19]. The inflammatory infiltrate is composed of lymphocytes, plasma cells, monocyte/macrophages, and sometimes significant numbers of eosinophils [3, 13] (Fig. 23.1). Eosinophils usually make up 2–10 % of the infiltrating cells [20, 73, 79]. Granuloma formation may be seen with palisading macrophages with an epithelioid character [3, 13, 69, 80–82]. Granulomatous interstitial nephritis may be seen in other forms of AIN such as sarcoid and TINU Syndrome [81]. “Tubulitis,” defined as an invasion of the tubules by lymphocytes and other cells, is a characteristic feature of AIN and may be found in as many as 80 % of biopsies of AIN patients [3, 13, 67]. This more frequently involves the distal nephron [83]. In classic AIN the presence of tubulitis and only focal tubular necrosis contrasts with the more extensive tubular damage and sparser interstitial infiltrates found in acute tubular necrosis. Nevertheless, the distinction between these entities is not always clear-cut. The glomerular and vascular compartments are generally spared in medication-related AIN with the exception of disease related to NSAIDs.
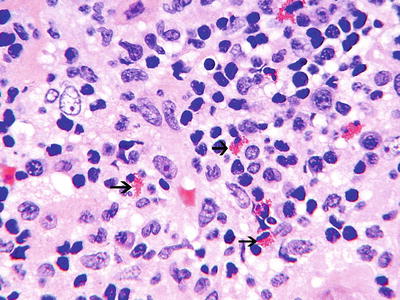
Fig. 23.1
Interstitial nephritis, showing a predominantly mononuclear infiltrate with few eosinophils (black arrows) (H&E ×40)
Immunofluorescence microscopy is usually negative in medication-related AIN with neither complement nor immunoglobulins found along the tubules or within the interstitium [3, 18, 19, 68]. Rarely, there is linear or granular staining for IgG and C3 along TBMs [18, 59]. Likewise, by electron microscopy only rarely have TBM electron dense deposits been noted [68, 84].
The inflammatory infiltrates in most cases of NSAID-related AIN are characterized by lymphocytes and plasma cells and less frequently by eosinophils [27, 28, 60, 61]. Most cases associated with the nephrotic syndrome have shown the lesion of minimal change disease, with normal glomeruli by light microscopy, negative immunofluorescence, and EM only effacement of the foot processes [27, 28, 60, 61]. Others cases have been associated with a membranous pattern of glomerular injury [85–87].
In AIN associated with infections in addition to edema and interstitial inflammation, neutrophils may be more prominent [35–40]. Although in AIN associated with systemic or glomerular disease the pathology of the interstitium varies, a patchy interstitial inflammatory infiltrate is common to all [1]. In immune complex diseases, such as lupus and cryoglobulinemia, there may be granular deposits of immunoglobulin and complement along the TBMs [54, 55]. In idiopathic hypocomplementemic interstitial nephritis, there are massive tubulointerstitial immune deposits [58]. The renal involvement in Sjogren’s syndrome typically consists of a lymphocytic interstitial infiltration without immune deposits [52, 53]. In sarcoidosis a granulomatous interstitial nephritis is found in up to 20 % of patients [50, 51]. In TINU syndrome the renal pathology usually shows a variable inflammatory infiltrate of eosinophils, lymphocytes, and plasma cells occasionally with granuloma formation [56, 57].
Clinical Manifestations and Laboratory
The classic picture of medication-related AIN has best been described for the beta-lactam antibiotics, the penicillins and cephalosporins. Several hundred cases have been reported occurring in all decades of life and in both genders [18–21]. Although the duration of therapy often lasts for many weeks, the dose of the antibiotic has not been excessive [18]. The cardinal systemic features are the hypersensitivity triad of rash (30–50 %), fever (75–80 %), and peripheral blood eosinophilia (80 %) [18–21, 72]. However, at the time of onset of renal insufficiency, fewer than one-third of patients have the complete triad [18]. The rash is usually a classic drug eruption with maculopapular erythematous lesions on the trunk and upper extremities. The secondary fever spike often occurs 2–3 weeks into the course of the antibiotic therapy. Less common features are loin pain, arthralgias, lymphadenopathy, and other systemic organ involvement, e.g., hepatitis.
Urinary output may be maintained and nonoliguric acute kidney injury is more common than the oliguric form. Urinary findings include mild proteinuria, pyuria, and leukocyte casts. Hematuria is present in over 90 % of cases and gross hematuria is common in children. Large amounts of proteinuria and erythrocyte casts in the urine sediment are rare and can usually be explained by coincidental glomerular pathology [1–10, 88]. With the exception of NSAID-induced disease, only a few cases of AIN have been associated with glomerular pathology [32, 89, 90]. The finding of eosinophils in the urinary sediment, eosinophiluria, is both frequent and helpful in establishing a diagnosis [20, 21]. In a study of patients with acute kidney injury, all nine patients with methicillin AIN had eosinophiluria on Wright stain of the urinary sediment, while none of 43 other patients had this finding [20]. In a second study, 10 of 11 patients with AIN had eosinophiluria versus none of 30 patients with ATN, none of 10 with acute pyelonephritis, and only 1 of 15 with acute cystitis [91]. In this study the Hansel stain was superior to the Wright stain in confirming eosinophiluria. The precise number of urinary eosinophils diagnostic of AIN has been debated. However, one study suggested eosinophiluria of over 5 % of the total urinary leukocytes to be strongly suggestive of AIN [79]. However, urinary eosinophils are also commonly found in some cases of rapidly progressive glomerulonephritis, with renal atheroembolic disease and with acute prostatitis [79].
The kidneys in AIN typically are found to be normal sized or enlarged with increased cortical echogenicity by ultrasonography [1–10, 72]. Gallium scanning in drug-related AIN may show intense diffuse bilateral uptake of the gallium nuclide. Although this is rarely found in ATN, positive gallium scintigraphy has been noted in acute bacterial pyelonephritis and acute glomerulonephritis and even in several patients with minimal change nephrotic syndrome [92–94]. The urinary sodium and the fractional excretion of sodium have been elevated (>60 mEq/L and >1 % respectively) in only some studies [95]. Hypocomplementemia is rare [18].
Medications other than the beta-lactam antibiotics may be associated with AIN with similar or unique clinical features. In some patients there are no extrarenal manifestations accompanying the renal insufficiency [1–10]. Both sulfonamides and trimethoprim-sulfamethoxazole have been associated with classic AIN with the full hypersensitivity triad [1–10, 18]. AIN associated with the use of the quinolone antibiotics has been reported frequently [96–98]. The picture often has both classic clinical and histopathologic features.
The pattern seen with rifampin is unique [99–101]. With either intermittent or discontinuous therapy or on rechallenge with rifampin, patients have experienced sudden onset of fever, flank pain, hematuria and acute kidney injury. Some cases are associated with myalgias, hemolysis, and thrombocytopenia. Renal biopsy reveals a spectrum of findings from classic AIN to typical acute tubular necrosis. In rare cases, continuous therapy with rifampin has produced AIN with the presence of nephrotic range proteinuria [89]. Other antimicrobials associated with AIN infrequently or in a less well-documented fashion are listed in Table 23.2.
Although diuretics typically produce renal insufficiency by causing intravascular volume depletion, a number of them including thiazides and chlorthalidone, furosemide, ethacrynic acid, and ticrynafen have rarely produced AIN [102, 103], which typically has occurred in patients with prior renal disease and often displays the classic hypersensitivity features of rash, fever, and eosinophilia.
While the NSAIDs are commonly associated with renal and electrolyte disturbances due to prostaglandin inhibition [29, 60, 61], when associated with AIN they present unique clinical features, including development in the older population receiving these medications and after a prolonged prior exposure to the drugs (months to years) [1, 10, 30, 60, 61]. The hypersensitivity triad of rash, fever, and eosinophilia is uncommon as are hematuria and eosinophiluria even when interstitial eosinophilia is found on renal biopsy [29]. Finally, AIN caused by the NSAIDs has often been associated with the nephrotic syndrome usually of the minimal change disease pattern [29, 30, 60, 61, 104]. The onset of the acute renal failure and the nephrotic syndrome are often concurrent, and both typically remit within weeks of discontinuing the NSAID. Membranous nephropathy and very rarely focal segmental glomerulosclerosis have also been noted in patients taking NSAIDs [85–87]. NSAID-associated AIN may be associated with more frequent permanent renal dysfunction perhaps as a result of its paucity of clinical findings and its insidious nature [1, 6, 13].
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


