Julia Spencer Barthold, MD
Embryology of Testicular Development and Descent
Testicular Development
The human gonadal ridge containing somatic and germ cells is first identified on the medial urogenital ridge at 32 days after ovulation and is indistinguishable between males and females (Hanley et al, 1999). Primordial germ cells migrate from the yolk sac and differentiate into gonocytes during the 5th week of gestation. Expression of the earliest testis (Sertoli cell)-specific genetic markers, SRY (sex-determining region Y) and SOX9 (SRY-box 9), begins at between 41 and 44 days (Hanley et al, 2000) followed by the histologic appearance of Sertoli cells and testicular cords in male fetuses by 9 weeks of age (Ostrer et al, 2007). Migrating mesonephric cells contribute to testis cord formation as well as testicular endothelial cells (Cool and Capel, 2009) The germ cell marker, c-KIT, first identified at 7 weeks of age, is present in an increasing number of cells until 12 weeks, with subsequent reduced expression as maturation to intermediate germ cells progresses (Ostrer et al, 2007). By the third trimester, a third stage of germ cells, prespermatogonia, predominates within the testis. Leydig cells can be identified by cell-specific staining by 9 weeks and by peritubular myoid cells by 12 weeks of age.
Gonadal differentiation is more complicated than the original concept of ovarian development as the default pathway because emerging evidence suggests that activation of specific gene pathways is required for both male and female sex determination in mice (Blecher and Erickson, 2007; Cool and Capel, 2009). Sry has been considered the master male gene, but other genes required for gonadal determination act upstream of Sry and its downstream effects on Sox9 expression are indirect via derepression of an intermediate gene, likely Rspo1 (R-spondin homolog). Testicular determination is directly dependent on activation of Sox9 and Fgf9 (fibroblast growth factor-9), whereas ovarian determination is dependent on activation of Rspo1 and Wnt4 (wingless-related MMTV integration site-4) and the appropriate dosage of the X-linked Nr0b1 (nuclear receptor subfamily 0, group B, member 1, also known as Dax1) gene.
Hormonal function of the human fetal testis is critical for masculinization of the reproductive tract and testicular descent. Fetal Leydig cell development is divided into three phases: a proliferation and differentiation phase between 7 and 14 weeks’ gestation, a maturation phase until 18 weeks, and an involution phase that continues until term (Svechnikov and Soder, 2008). Synthesis of testosterone by fetal Leydig cells starts as early as 6 to 7 weeks’ gestation and appears to be initially independent of gonadotropin stimulation. However, placental human chorionic gonadotropin (hCG) stimulates a peak in androgen production at 14 to 16 weeks’ gestation and the testis then becomes responsive to fetal luteinizing hormone (LH). Insulin-like 3 (INSL3) of Leydig cell origin is measurable in human amniotic fluid as early as 13 weeks’ gestation (the earliest time point studied) and peaks at 15 to 17 weeks (Anand-Ivell et al, 2008; Bay et al, 2008). Fetal Sertoli cells produce antimüllerian hormone (AMH; also called müllerian-inhibiting substance [MIS]) soon after they differentiate; the human fetal müllerian duct is responsive to AMH before week 8 of gestation, and the process of regression occurs between 9 and 10 weeks (Josso et al, 2006). Evidence from animal models and indirect clinical evidence suggests that INSL3 and testosterone are the key hormones required for testicular descent.
Steroid hormones exert their effects via sex steroid receptors in the reproductive tract and testis. Recent studies show that androgen receptor (AR) and estrogen receptor-β (ERβ) are expressed in the undifferentiated gonad at 7 weeks of age, although their role in testicular development is unclear (Shapiro et al, 2005; Boukari et al, 2007). AR is present primarily in peritubular myoid cells and in some Leydig and interstitial cells but absent in Sertoli cells during the fetal period. Because androgens inhibit AMH production by Sertoli cells, the absence of an AR in these cells during fetal life allows unfettered expression of AMH during this period (Boukari et al, 2009). ERβ localization is most extensive, being observed in germ, peritubular myoid, Sertoli, and some Leydig cells by 13 weeks and decreasing by 22 to 24 weeks, whereas expression of estrogen receptor-α (ERα) is limited (Shapiro et al, 2005) or absent (Boukari et al, 2007) in the human fetal testis. Concomitant expression of aromatase during the same time frame suggests that locally produced estrogen may play a role in testicular development.
Significant changes in reproductive hormone secretion and testicular development occur in the neonatal period (Grumbach, 2005). Fetal Leydig cells regress after birth, followed by emergence of a neonatal Leydig cell population at 2 to 3 months of age (Prince, 2001) and a peak in testosterone and INSL3 levels (Bay et al, 2007) that follows a surge in serum gonadotropin levels. As postnatal hormonal levels wane, the fetal Leydig cells either degenerate or regress to immature Leydig/interstitial cells that are partially differentiated and less responsive to LH. In the first months of life, intratubular gonocytes migrate to the basement membrane and differentiate into type A spermatogonia, which comprise a subset of spermatogonial stem cells (Culty, 2009). Sertoli cell proliferation occurs in the first year of life and is a major determinant of ultimate testicular size (Sharpe et al, 2003). The characteristics of testicular growth during childhood vary depending on the method of analysis. Main and colleagues reported significant growth of normal testes between birth and 3 months of age based on ultrasound measurements and on significantly larger testicular size in Finnish as compared with Danish boys at birth and 3 and 18 months, a difference attributable to genetic and/or environmental effects (Main et al, 2006b). Kuijper and coworkers observed a peak in testicular ultrasound volume at 6 months of age followed by a decline to baseline (Kuijper et al, 2008). In contrast, in an autopsy series, Berensztein and associates noted doubling of testicular weight during the first 3 weeks of life with slowed growth thereafter, suggesting that major growth occurs prior to the postnatal hormone surge and a gonadotropin-independent mechanism (Berensztein et al, 2002). This growth was associated with a rate of proliferation exceeding the rate of apoptosis in Leydig, germ, and Sertoli cells before the second month of age with increased apoptosis thereafter and little subsequent growth of the testes before age 6. In their autopsy series of older children, Müller and Skakkebaek (1983) reported a 2.6-fold increase in testicular size between the first year of life and age 10, with a significant increment between all age groups (birth to 1, 1 to 5, and 5 to 10 years) in both size and tubular length. Overall, the data suggest a major increment in testicular growth during the neonatal period, but the factors responsible for age-specific cellular proliferation, gonocyte differentiation, and testicular growth are not clearly defined.
Gubernacular Development and Testicular Descent
The human indifferent gonad develops adjacent to and becomes suspended from the mesonephros, which has replaced the pronephros by 5 weeks’ gestational age (Lemeh, 1960). A rudimentary cranial mesonephric ligament connects to the diaphragm, disappearing by 13 weeks as the mesonephros regresses (Barteczko and Jacob, 2000) (Fig. 132–1). There is no direct connection between the gonad and this ligament; therefore no “cranial gonadal ligament” exists in humans as noted in other species. Using fixed sections, Barteczko and Jacob observed an “inner” or transabdominal descent of the human testis from a position at vertebral levels C7-T8 to T9-L3 between 5 and 7 weeks’ gestation and to the sacral level by 10 weeks; using dissections, others described the testis at the level of L4 by the 7th week (Lemeh, 1960) or always “close to the groin” (Wyndham, 1943). Caudal movement of the ovary is prevented by the ovarian ligament and development of the müllerian ducts.
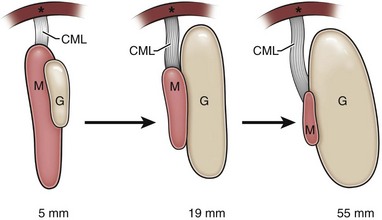
(From Barteczko KJ, Jacob MI. The testicular descent in human: origin, development and fate of the gubernaculum Hunteri, processus vaginalis peritonei, and gonadal ligaments. Adv Anat Embryol Cell Biol 2000;156:III-X, 1–98.)
Barteczko and Jacob (2000) described five major phases of testicular descent in the human fetus (Fig. 132–2):
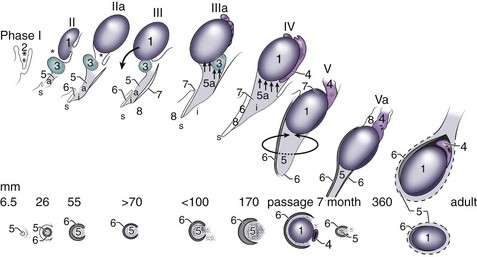
(From Barteczko KJ, Jacob MI. The testicular descent in human: origin, development and fate of the gubernaculum Hunteri, processus vaginalis peritonei, and gonadal ligaments. Adv Anat Embryol Cell Biol 2000;156:III-X, 1–98.)
The careful in-situ anatomic observations by Barteczko and Jacob (2000) throughout gestation help clarify some aspects of human testicular descent. For example, transabdominal movement of the testis occurs before sexually dimorphic changes in the gubernaculum and is presumably not a male hormone–specific event. The subsequent male-specific swelling of the gubernaculum parallels the peak in Leydig cell secretion of testosterone (14 to 16 weeks) and INSL3 (15 to 17 weeks). The genitofemoral nerve accompanies and precedes the gubernaculum and developing processus vaginalis in both sexes beginning at an early stage (7 weeks), and both smooth (abdominal) and skeletal (interstitial and subcutaneous) muscle fibers occur within the three segments of the gubernaculum. Both muscle types were also observed by some (Wyndham, 1943; Lemeh, 1960) but not others (Heyns, 1987; Costa et al, 2002; Niikura et al, 2008) observers. However, studies of the in-situ gubernaculum beginning at 20 weeks clearly show the presence of peripherally and centrally located striated muscle bundles (Barteczko and Jacob, 2000; Niikura et al, 2008) and infiltration of the gubernaculum by the genital branch of the genitofemoral nerve (Tayakkanonta, 1963). Controversy exists as to whether these skeletal cremaster muscle fibers are of similar origin and/or originate from abdominal wall musculature or from within the gubernaculum itself (van der Schoot, 1996; Barteczko and Jacob, 2000; Niikura et al, 2008). The processus vaginalis and cremaster muscle may facilitate transit of the testis and gubernaculum, but their exact role remains unknown.
Swelling of the gubernaculum is critically important to allow enlargement of the inguinal canal and testicular passage and is the result of both cellular proliferation and production of extracellular matrix (Heyns, 1987). Once the canal is created, unknown mechanical factors trigger a typically rapid transinguinal passage of the testis. The gubernaculum is unattached distally during and after transinguinal passage (Fig. 132–3). Settling of the gubernaculum and testis into the preformed scrotum is gradual. The scrotum itself develops from genital swellings that are first visible at 7 weeks and fuse by 10 to 12 weeks in response to circulating dihydrotestosterone. Passage of the testis into the inguinal canal rarely occurs prior to week 22 in the human fetus, and the majority of testes are scrotal after week 27; time spent in the inguinal canal seems to be limited for the majority of human fetal testes (Heyns, 1987; Sampaio and Favorito, 1998). Early innervation of the gubernaculum and the importance of the genitofemoral nerve in testicular descent in rodents, as well as the rapidity of transinguinal passage, suggest a neuromuscular mechanism.
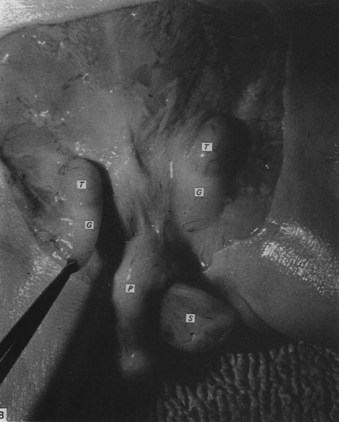
(From Heyns CF. The gubernaculum during testicular descent in the human fetus. J Anat 1987;153:93–112.)
Regulation of Testicular Descent
Direct evidence for hormonal control of testicular descent is based entirely on studies in animal models, primarily rodents. The relevance of rodent data to humans is controversial because of anatomic differences, which in mice and rats include a less prominent interstitial gubernaculum, a cranial gonadal ligament, an elongated cord connecting the gubernaculum to the epididymis, a well-developed intrinsic cremaster muscle located in the outer layer of the gubernaculum, an absent inguinal canal, and failure of closure of the processus vaginalis (Wensing, 1988). However, some observers argue that the similarities between species are sufficiently strong to warrant translational studies of these models (van der Schoot, 1996; Harnaen et al, 2007).
Rodent studies provide strong evidence that testicular INSL3 and androgen signaling pathways are the primary contributors to gubernacular development and testicular descent (Zimmermann et al, 1999; Adham et al, 2000; Overbeek et al, 2001). According to a paradigm developed by Hutson, testicular descent occurs in two phases—transabdominal and transinguinal—regulated by INSL3 and by androgens via the genitofemoral nerve, respectively (Hutson and Hasthorpe, 2005). However, animal models show that INSL3 and androgen synergistically stimulate cellular proliferation and growth of the gubernaculum (Adham et al, 2000; Emmen et al, 2000; Kubota et al, 2002) and that both INSL3 binding and androgen receptor expression are present before and during the swelling phase of the fetal rat gubernaculum (Koskimies et al, 2003; McKinnell et al, 2005; Staub et al, 2005). Moreover, INSL3 and testosterone levels peak after week 12 in the human fetus, concomitant with gubernacular swelling, although transabdominal testicular descent occurs earlier, prior to 10 weeks’ gestation. In mice, INSL3 signaling promotes development of the processus vaginalis, and androgen signaling causes regression of the cranial gonadal ligament (Adham et al, 2002; Koskimies et al, 2003; Adham and Agoulnik, 2004), a structure that does not exist in the human fetus. Clues about other gene pathways that participate in the process of testicular descent are provided by known transgenic mouse models and human syndromes with phenotypes that include cryptorchidism (Barthold, 2008; Foresta et al, 2008). Transgenic deletion of either of two homeobox genes, Hoxa10 and Hoxa11, is associated with isolated cryptorchidism in mice. Hoxa10 appears to function independently of gonadotropin or INSL3 signaling (Feng et al, 2006). At present, little is known of the role of these and other gene candidates in the process of testicular descent.
A role for the genitofemoral nerve in guiding and/or promoting passage of the gubernaculum is supported by rodent studies of Hutson and colleagues, who propose that release of calcitonin gene-related peptide (CGRP) from the nerve stimulates development and function of the gubernaculum (Yamanaka et al, 1993; Chan et al, 2009). Studies of rats with inherited cryptorchidism suggest that innervation and/or muscle development within the gubernaculum is disrupted (Hrabovszky et al, 2001; Barthold et al, 2008). While a role for CGRP release from the genitofemoral nerve in human fetuses remains undefined, it is clear that the human gubernaculum contains and is surrounded by muscle and is innervated by the nerve beginning as early as at 7 weeks’ gestation (Lemeh, 1960; Tayakkanonta, 1963; Barteczko and Jacob, 2000; Niikura et al, 2008).
Key Points
Embryology of Testicular Development and Descent
Cryptorchidism (Undescended Testis)
Cryptorchidism is one of the most common disorders encountered in pediatric urology and has been studied extensively. Despite this, knowledge of the causes and prediction of the consequences of the disease remains limited. Moreover, the quality of evidence that is used to support clinical guidelines for diagnosis and treatment is surprisingly low (Gapany et al, 2008; Turner, 2009).
Definitions
Differing terminology has been used to describe patients with nonscrotal testes; use of the following definitions is recommended. Normal scrotal position has been defined as positioning of the midpoint of the testis at or below the midscrotum (Wohlfahrt-Veje et al, 2009). Although “high scrotal testes” are not routinely considered undescended by most clinicians, they have been included in the definition of undescended testis in some epidemiologic studies (Sijstermans et al, 2008). This is likely a heterogeneous group that includes stable descended testes that reside above the scrotal midpoint and undescended “gliding” testes (Hack et al, 2007), which are not stable. Undescended testis is the absence of one or both testes in normal scrotal position and during initial clinical evaluation may refer to palpable cryptorchid testes or to nonpalpable testes, which are either cryptorchid or absent. Most absent testes are vanishing, present initially in development but are lost owing to vascular accident or torsion unilaterally (monorchia) or, very rarely, bilaterally (anorchia) (Abeyaratne et al, 1969). Agenesis refers to a testis that was never present and therefore associated with ipsilateral müllerian duct persistence. Congenital cryptorchidism refers to testes that are extrascrotal at birth, and recurrent cryptorchidism is when testes descend spontaneously postnatally but subsequently return to a nonscrotal position. The terms testicular ascent or acquired cryptorchidism are used in situations when cryptorchid testes are documented as scrotal at a previous examination without intervening inguinal surgery. Secondary cryptorchidism and testicular retraction apply to testes that are suprascrotal after inguinal hernia repair and as a complication of orchidopexy, respectively. Testicular malposition after hernia repair is due to either postoperative scarring or primary maldescent. Retractile testes are scrotal testes that retract easily out of the scrotum but can be manually replaced in a stable scrotal position and remain there at least temporarily. Testes that are significantly retractile, that is, those that rarely remain in a stable scrotal position (spontaneously or with manipulation) and/or are located at rest in the high scrotum, may or may not be truly undescended.
Epidemiology and Pathogenesis
Cryptorchidism is one of the most common congenital anomalies, occurring in 1% to 4% of full-term and 1% to 45% of preterm male neonates (Sijstermans et al, 2008). It is a component of over 390 syndromes (Winter-Baraitser Dysmorphology Database, http://www.lmdatabases.com); the most common are listed in recent reviews (Foresta et al, 2008; Virtanen and Toppari, 2008). The majority of cases are isolated, with the ratio of nonsyndromic to syndromic cryptorchidism reported as greater than 6 : 1 in a large cohort (Boyd et al, 2006). In a small percentage of syndromic cases associated with deficiency or insensitivity of hypothalamic-pituitary-gonadal (HPG) axis hormones, the etiology of testicular maldescent is known. The pathogenesis of isolated cryptorchidism remains largely unknown but is most likely multifactorial, involving both genetic and environmental risk factors.
Nonsyndromic Congenital Cryptorchidism
Studies of the prevalence of isolated cryptorchidism at birth are complicated by confounding factors that include subjectivity of the examination and differences in the definition of undescended testis (inclusion/exclusion of high scrotal testes) and in study populations and study design (Sijstermans et al, 2008). Although most studies support a prevalence at birth of 2% to 4% and at 3 months of age of 1% to 2%, this varies geographically, with frequency as high as 8% in some studies (Virtanen and Toppari, 2008), supporting the possibility of an increase over time. However, other data suggest that country-specific trends are not increasing (Abdullah et al, 2007; Cortes et al, 2008; Bonney et al, 2009) and overall there do not appear to be reproducible trends in prevalence (Sijstermans et al, 2008). Perinatal risk factors associated with cryptorchidism include prematurity, low birth weight/small for gestational age, breech presentation, and maternal diabetes (Damgaard et al, 2008; Virtanen and Toppari, 2008).
The reported frequency of spontaneous testicular descent after birth also appears to vary substantially between studies, likely owing to similar confounding factors. Of 95 patients with cryptorchidism referred to a urology practice when younger than 6 months of age, 16% had spontaneous testicular descent versus 0% of those presenting after 6 months; in contrast, previous studies reported a 50% to 75% rate of spontaneous postnatal testicular descent (Wenzler et al, 2004). Suomi and associates (2006) noted spontaneous descent in significantly more Danish (68%) than Finnish (45%) boys at 3 months of age, likely owing to increased severity of the disease in the latter group. Similarly, in a recent longitudinal study, extrascrotal testes were much less likely to descend by 1 year of age (50%) than high scrotal testes defined as cryptorchid at birth (87.5%) (Acerini et al, 2009). The long-term risk of recurrent cryptorchidism was not determined in these studies and is difficult to ascertain because of short follow-up. Spontaneous descent is more likely and may occur later in premature infants.
Retractile and Acquired Undescended Testes
In the past, cryptorchidism was considered a congenital anomaly identifiable at birth, with spontaneous descent possible during the postnatal period. However, since first reported more than 30 years ago (Myers and Officer, 1975), substantial evidence indicates that the diagnosis of cryptorchidism may be “acquired,” that is, made in cases of apparent full descent at birth or after spontaneous descent of a cryptorchid testis (Barthold and Gonzalez, 2003; Taghizadeh and Thomas, 2008). Except for diagnosis at an average age of 8 to 9 years and lower testicular position, other characteristics of testicular “ascent” or “acquired” cryptorchidism are similar to those observed in cases of congenital cryptorchidism. The reason for a later diagnosis remains unknown; theories include presence of a fibrous remnant of the processus vaginalis that tethers or foreshortens the cord over time or mobility of the testis within an open sac. However, it is most likely that the testis is incompletely descended from birth, because many are localized within the superficial inguinal pouch (Barthold and Gonzalez, 2003). These testes may be highly mobile and initially appear descended until somatic growth results in relative widening of the distance between testis and scrotum (Redman, 2005; Agarwal et al, 2006).
A diagnosis of cryptorchidism beyond the neonatal period may be more likely in boys with retractile testes. In an unselected population, the testis was reported to be suprascrotal on initial examination in up to 30% of boys at 4 years of age but intrascrotal in 100% by age 12 (Farrington, 1968). Both prospective and retrospective studies of the natural history of retractile testes support the concept that a subset of these becomes undescended over time. However, a selection bias exists in studies of the risk of cryptorchidism in boys with retractile testes, because those referred for follow-up by specialists are those with the most severe retractility. For example, Wyllie (1984) prospectively studied a cohort of 100 boys with unilateral retractile testis and identified 64 cases in which testicular position, as documented by the distance between the pubic tubercle and midtestis, and/or testicular size, as estimated by orchidometer, was reduced after 5 years of follow-up. Orchidopexy was performed in 45 of these cases, although specific documentation of testicular position and size as indications for surgery were not reported. Findings of retrospective case series also suggest that cryptorchidism is ultimately diagnosed in about 33% of boys with significantly retractile testes followed longitudinally by the same observer(s) (La Scala and Ein, 2004; Agarwal et al, 2006). In contrast, only 7% of 172 boys followed a mean of 26 months required orchidopexy in a more recent study (Stec et al, 2007). These data suggest that a significant minority of retractile testes become suprascrotal over time. Careful, often serial, physical examination may be necessary to accurately determine testicular position and need for intervention.
Two recent longitudinal population-based cohort studies have provided some preliminary information regarding the risk of acquired cryptorchidism in the general population during childhood (Acerini et al, 2009; Wohlfahrt-Veje et al, 2009). In a study from the United Kingdom, 742 infants were followed with serial examinations for 2 years after birth with complete follow-up in 326. The prevalence of extrascrotal testes was 2.7% at birth (27% nonpalpable, surgical findings not reported); and 0.2%, 1.8%, 0.3%, and 0% of testes in the studied population became extrascrotal at 3, 12, 18, and 24 months, respectively. Including high scrotal testes, a total of 5.7% were identified as cryptorchid at birth and 1% to 4% rose to a high scrotal position with each follow-up visit. As expected, suprascrotal testes in neonates were much less likely to descend spontaneously (41%) than high scrotal testes (84%). In a series from Finland, cryptorchidism was present at birth in 94 of 1072 boys (0.9%) and 509 of the noncryptorchid boys were followed at 6- to 18-month intervals until 4.5 to 10 years of age. Of those observed, seven testes became suprascrotal and five became high scrotal, with the change in position occurring during the first 36 months of life. These studies suggest regional differences in both congenital and acquired cryptorchidism, but both suggest a low risk that scrotal testes will become suprascrotal in the first few years of life. However, cases diagnosed during childhood comprise a significant number of those requiring orchidopexy and yearly well-child screening is indicated.
Genetic Susceptibility
Genetic studies of cryptorchidism suggest that the disease is heritable but that susceptibility is likely polygenic and multifactorial. Clustering of cryptorchidism has been reported in a number of families affecting multiple individuals in the same generation and variable phenotype (Minehan and Touloukian, 1974; Pardo-Mindan et al, 1975; Czeizel et al, 1981; Savion et al, 1984). Extended pedigrees were not usually examined, but autosomal dominance with reduced penetrance was most often cited as the probable mode of inheritance. Population case-control studies also support genetic contribution to the disease. Familial aggregation suggesting moderate genetic risk was reported in a recent large cohort study of more than 1 million male births in the Netherlands based on extensive hospital registry data (Schnack et al, 2008). Recurrence risk ratio (RR) was 10.1 in twins, 3.5 in brothers, and 2.3 in offspring and were significantly higher in maternal than in paternal half-brothers. Previous smaller studies reported a 5- and 7- to 10-fold increased risk of cryptorchidism in fathers and brothers, respectively, of affected as compared with unaffected subjects (Czeizel et al, 1981; Jones and Young, 1982; Elert et al, 2003). These and another recent study (Jensen et al, 2010) support the possibility that maternal factor(s), or X-linked risk alleles, influence expression of the disease.
Based on mouse models, INSL3, its receptor, relaxin/insulin-like family peptide receptor 2 (RXFP2), HOXA10, and HOXA11 were considered the most likely candidate genes for human nonsyndromic cryptorchidism. However, case-specific DNA coding variants of INSL3 and RXFP2 are reported in only 0.6% to 1.8% and 1.6% to 2.9% of persistently cryptorchid males, respectively, and in-vitro assays suggest that few of these appear to be functionally significant (Foresta et al, 2008). Notably, apparent mutations in these genes are associated with a range of phenotypes encompassing unilateral/bilateral and persistent/spontaneously resolving presentations and apparently deleterious mutations may exist in normal family members, as recently noted for an INSL3 mutation showing reduced receptor activation (El Houate et al, 2007). A nonsynonymous polymorphism of the RXFP2 gene, T222P, was considered a strong etiologic candidate for cryptorchidism based on absence in normal controls and in-vitro studies showing curtailed cell membrane localization and activation of the abnormal receptor (Bogatcheva et al, 2007). However, cases and controls in a different European population subset were found to have a similar frequency of the T222P allele (Nuti et al, 2008). Although these data suggest that mutations of INSL3 and RXFP2 are infrequent in cases of cryptorchidism, it is possible that noncoding variants that alter expression levels of the protein have yet to be identified.
Analysis of other potential candidate genes for human cryptorchidism has failed to yield reproducible results. HOXA10 mutations reported by Kolon and colleagues (1999) were not confirmed in additional series, nor were mutations identified in HOXA11 (Bertini et al, 2004; Wang et al, 2007). A specific haplotype of the gene encoding ERα (ESR1) has not shown consistent association with cryptorchidism in three case-control series (Yoshida et al, 2005; Galan et al, 2007; Wang et al, 2008). Similar varying results have been reported in studies of trinucleotide repeat number in exon 1 of the androgen receptor, although more recent work suggests a possible association with cryptorchidism (Ferlin et al, 2007). Microdeletions of the Y chromosome have also been studied in infertile men with and without a history of cryptorchidism, and no consistent association was observed (reviewed in Gurbuz et al, 2008). A specific polymorphism of the steroidogenic factor 1 (SF1) gene that encodes a transcription factor that regulates expression of INSL3, RXFP2, and steroidogenesis genes was recently reported to exist more commonly in cryptorchid Japanese males (Wada et al, 2006). Failure to identify consistent differences in genomic variants for many of these candidates may be influenced by confounding factors such as race and by insufficient sample size. Although multiple genetic variants likely contribute to the risk of nonsyndromic cryptorchidism, most remain unknown at this time.
Environmental Risk Factors
Sharpe and Skakkebaek (2008) proposed that nonsyndromic cryptorchidism is one component of the “testicular dysgenesis syndrome,” a constellation of reproductive abnormalities including testicular cancer, hypospadias, and reduced sperm counts associated with exposure to antiandrogenic and/or estrogenic environmental endocrine-disrupting chemicals (EDCs). However, some question both the existence of testicular dysgenesis syndrome in humans, because of lack of epidemiologic association between these abnormalities, and the evidence for environmental etiology (Akre and Richiardi, 2009). The concern for a link between EDCs and cryptorchidism arose because of a reported increased risk of cryptorchidism after maternal exposure to diethylstilbestrol (DES) (Gill et al, 1979). This association was observed in rodents but was not confirmed in other clinical case-control studies (Leary et al, 1984); however, in a recent large cohort study of 1197 men previously exposed to DES and 1038 unexposed men, the relative risk (RR) of cryptorchidism was 1.9 overall (confidence interval [CI] 1.1 to 3.4). When data were subdivided based on gestational age of less than 11 weeks and exposure greater than or equal to 5 g, the relative risks were 2.9 (CI 1.6 to 5.2) and 3.2 (CI 1.7 to 6.0), showing that early and significant exposure presents the clearest risks (Palmer et al, 2009). Data supporting a link between other EDCs and cryptorchidism are less clear. Animal models suggest a relationship between prenatal exposure to antiandrogenic EDCs and risk of cryptorchidism and other reproductive endpoints and suggest additive effects of chemical mixtures, although levels of exposure are much higher than applicable clinically (Rider et al, 2009). In humans, data supporting a correlation between exposure to antiandrogenic and/or estrogenic EDCs such as pesticides, flame retardants, and phthalates and occurrence of cryptorchidism are mostly indirect or suggestive (Weidner et al, 1998; Pierik et al, 2004; Damgaard et al, 2006; Main et al, 2006a, 2007; Fernandez et al, 2007; Krysiak-Baltyn et al, 2010). Strong correlations between single chemicals or classes of EDCs and risk of cryptorchidism may be difficult to demonstrate in view of the presumed heterogeneity of susceptibility to the disease, but the wide variety of EDCs to which humans are exposed may allow for synergistic effects that are difficult to measure. However, at present, epidemiologic data are suggestive but do not strongly support the theory that environmental chemicals increase susceptibility to cryptorchidism.
Testicular hormones are required for testicular descent; therefore, if defective hormone production and/or action contribute to the pathogenesis of cryptorchidism, hormone deficiency may persist postnatally. A number of prospective studies of varying size and quality are available that report postnatal hormone levels in cryptorchidism; although some data suggest that the HPG axis is abnormal, the results are conflicting. In the first reported series of cryptorchid and control boys undergoing hormonal evaluation in the first few months of life, serum testosterone was lower in 7 of 17 (41%) persistently cryptorchid boys but comparable to control levels in 4 of 25 boys with spontaneous testicular descent (Gendrel et al, 1978). In a small series of 60-day-old bilaterally cryptorchid (9) and control (14) boys, Facchinetti and associates (1983) also observed reduced plasma testosterone levels but no difference in plasma LH between groups. Raivio and coworkers (2003) reported both significantly reduced plasma testosterone as well as nonmeasurable androgen bioactivity in 3-month-old cryptorchid boys (n = 45; 31% bilateral, 44% with spontaneous descent), but androgen bioactivity was measurable in only 21 of 55 controls.
Additional infant hormone studies did not show lower levels in cryptorchid infants (De Muinck Keizer-Schrama et al, 1988, Barthold et al, 2004), but more recent data from the Netherlands did show reduced testosterone levels in cryptorchid infants (Pierik et al, 2009). In the largest study to date, Suomi and colleagues (2006) showed geographic differences in serum inhibin-B and follicle-stimulating hormone (FSH) levels but no differences in testosterone levels in cryptorchid infants from Finland (n = 88, 36% spontaneous descent) and Denmark (n = 34, 68% spontaneous descent) as compared with 300 and 399 control boys, respectively. In a related cohort, INSL3 levels were reduced in cord blood but not in serum obtained at 3 months of age in persistently cryptorchid males (Bay et al, 2007).
Other studies of Sertoli/germ cell function as determined by serum assays in boys with cryptorchidism suggest that abnormalities may exist but data are inconsistent between studies. In a longitudinal study of 27 boys (mean age 4.8 years) undergoing orchidopexy, an increase in serum inhibin B was identified in a majority of cases 6 months postoperatively (Irkilata et al, 2004), but in another series of 62 boys (mean age 7.7 years) no differences were identified in inhibin B (or in serum LH, FSH, or testosterone) levels before or after hCG stimulation (Christiansen et al, 2002). AMH levels were measured in three prospective studies of boys with cryptorchidism as compared with age-matched controls. Lower AMH levels were seen in a subset of patients younger than 8 years of age included in a study of 104 cryptorchid boys (mean age 4 to 5 years) (Yamanaka et al, 1991) and also in a case-control study of 12-month-old boys (n = 20 per group) in whom levels did not change after orchidopexy (Demircan et al, 2006). However, in another study no differences in AMH levels were seen in 1- to 6-month-old cryptorchid (n = 43) versus control (n = 113) boys (Pierik et al, 2009).
These studies suggest that at least a subset of boys with cryptorchidism have measurable abnormalities in pituitary and/or gonadal hormone secretion during infancy without syndromic endocrine dysfunction. Inconsistent findings between studies are likely based on timing and type of assays, sample size, and heterogeneity based on age, severity, genetic background, and/or other interindividual differences. Lifestyle factors may also interfere with testicular descent and function via hormonal or nonhormonal effects; examples are the recent observation that smoking is associated with cryptorchidism in some case-control series (Thorup et al, 2006; Jensen et al, 2007) and the reduced expression of the Desert hedgehog (DHH) gene that is involved in Leydig cell differentiation in human fetuses (Fowler et al, 2008). Multiple genetic and environmental factors likely contribute to cryptorchidism, complicating identification of risk factors in case-control studies.
Syndromic Cryptorchidism
Undescended testes are frequently present in diseases associated with reduced androgen production and/or action, such as androgen biosynthetic defects, androgen insensitivity, Leydig cell agenesis, and gonadotropin deficiency disorders (Barthold et al, 2000; Foresta et al, 2008) that are typically associated with generalized failure of masculinization and are not reviewed here. AMH biosynthesis or receptor defects are also associated with cryptorchidism or transverse testicular ectopia (Josso et al, 2006). Gubernacular defects that are observed in these patients may be due to hindrance of descent by retained müllerian ducts (Barteczko and Jacob, 2000) and/or through loss of direct proliferative effects of AMH on the gubernaculum (Kubota et al, 2002). The frequency of Klinefelter syndrome (47,XXY) is 1.8% in large series of primarily nonsyndromic cryptorchidism but is more frequent when other anomalies, particularly hypospadias, are present (Sasagawa et al, 1996; Moreno-Garcia and Miranda, 2002). Other genomic rearrangements and trisomies, including Down syndrome (trisomy 21), are associated with cryptorchidism (Hadziselimovic, 1983).
Certain anomalies are associated with increased risk of cryptorchidism, many related to musculoskeletal, central nervous system (CNS), or abdominal wall/gastrointestinal defects. These include all cases of classic prune-belly (triad or Eagle-Barrett) syndrome; 80% of those of spigelian hernia (Durham and Ricketts, 2006); and 41% to 54% of cerebral palsy (Rundle et al, 1982; Cortada and Kousseff, 1984), 38% of arthrogryposis (Fallat et al, 1991), 15% of myelomeningocele (Ferrara et al, 1998), 16% to 33% of omphalocele, 5% to 15% of gastroschisis (Kaplan et al, 1986; Koivusalo et al, 1998), 19% of imperforate anus (Cortes et al, 1995b), 12% to 16% of posterior urethral valve (Krueger et al, 1980; Heikkila et al, 2008), and 6% of umbilical hernia (Kaplan et al, 1986) patients. Multisystem anomalies are often associated with omphalocele (80%) and prune-belly syndrome (45%), suggesting a syndromic etiology (Loder et al, 1992; Koivusalo et al, 1998). Depue (1988) also reported a significant association of cryptorchidism with CNS dysfunction, particularly cerebral palsy (RR = 34), low intelligence quotient (RR = 2.7), and hypotonia (RR = 3.6). Cortes and associates (1998) identified an association between renal and T10 to S5 spinal anomalies and cryptorchidism, with the affected testis on the same side as the renal anomaly in 90% of cases. In addition, cryptorchidism associated with syndromes and CNS malformations is more commonly bilateral (Cendron et al, 1993; Cortes et al, 1995a). These data support common origins of cryptorchidism and abnormalities of urogenital ridge, abdominal wall, lumbosacral spine, and CNS development.
Diagnosis and Classification
In large clinical series, the majority (75% to 80%) of undescended testes are palpable and 60% to 70% are unilateral; involvement of the right side is more common overall but less frequent in series of nonpalpable testes (Hadziselimovic, 1983; Cendron et al, 1993; Cortes et al, 2001; Giannopoulos et al, 2001). Position of undescended testes varies markedly with the population studied. In a comprehensive review of surgical patients, testes were abdominal in 34%, near the internal ring (“peeping”) in 12%, canalicular in 27%, and beyond the external ring in 27% (Docimo, 1995) whereas in a large single institutional series testes were abdominal in 3% to 10% of total cases, canalicular in 16% to 27%, with the majority distal to the external ring (Hadziselimovic, 1983; Moul and Belman, 1988; Cendron et al, 1993). In a large multi-institutional review of almost 40,000 European cryptorchid boys reported by Hadziselimovic (1983), 8% of testes were abdominal, 63% canalicular, 24% prescrotal, and 11% in the superficial inguinal pouch or ectopic. Moul and Belman classified all subinguinal testes with a lateral gubernacular attachment as ectopic (66% of their total cases).
Associated genital findings may warrant additional diagnostic studies that are best completed in the neonatal period. If both testes are nonpalpable, particularly if penile development is abnormal, karyotype and hormonal analyses are performed to rule out congenital adrenal hyperplasia and obviate the potential adverse effects of undiagnosed salt wasting. Hypospadias is associated with cryptorchidism in 12% to 24% of cases (Cendron et al, 1993; Moreno-Garcia and Miranda, 2002; Cox et al, 2008). If proximal hypospadias is present, chromosomal analysis is warranted because the frequency of abnormalities is high (32%) (Cox et al, 2008). Penile size may be small in boys with bilateral vanishing testes (also called testicular regression syndrome) or with hypogonadotropic hypogonadism, in whom undescended testes are most commonly inguinal. In these cases, measurement of testosterone, LH, and FSH levels in the first few months of life can facilitate identification of hormone deficiency or anorchia.
Palpable Testes
Difficulty in the clinical classification of cryptorchidism when the testis is palpable is related to both documentation of testicular position and differentiation of truly undescended from retractile testes, complicated by the fact that these entities may coexist. The gold standard for diagnosis remains careful examination of a child in several positions and confirmation of incomplete descent of the testis to a dependent scrotal position after induction of anesthesia. Prospective studies of intraobserver and interobserver variation show major differences in documentation of testicular position between examiners (Wit et al, 1987; Olsen, 1989). Olsen (1989) noted complete agreement between two examiners on scaled measures of testicular position and mobility in only 5 (13.5%, 95% CI 4.5% to 28.8%) of 37 boys. Cendron and coworkers (1993) reported that preoperative testicular position correlates poorly with intraoperative findings. Variation in observed testicular position preoperatively and postoperatively may influence assessment of prognosis and outcome in boys with cryptorchidism.
Nonpalpable Testes
When a testis is nonpalpable, possible clinical findings at surgery include (1) abdominal or transinguinal “peeping” location (25% to 50%), (2) complete atrophy (“vanishing” testis, 15% to 40%), and (3) extra-abdominal location but nonpalpable due to body habitus, testicular size, and/or limited cooperation of the patient (10% to 30%) (Cendron et al, 1993; Cisek et al, 1998; Kirsch et al, 1998; Radmayr et al, 2003; Patil et al, 2005). If both testes are nonpalpable and not distal to the internal inguinal ring in a genetic male, at least 95% are abdominal with bilateral vanishing testis occurring rarely (Cendron et al, 1993; Moore et al, 1994). If neither vas nor spermatic artery is found at the time of laparoscopy, laparoscopic or surgical dissection of the paravesical area and retroperitoneum up to the level of the kidney is required to exclude the presence of a testis. Pararenal or other abdominal testes may be associated with multicystic dysplastic or absent ipsilateral kidneys and/or nonunion of the testis and epididymis (Zaccara et al, 2004; Foley et al, 2005; Kim et al, 2005).
The etiology of vanishing testis is most likely torsion or vascular accident occurring after completion of genital masculinization but before fixation of the testis in the scrotum. Evidence supporting this etiology includes the presence of hemosiderin in remnant testicular “nubbins” excised at surgery (Turek et al, 1994) and reported cases of contralateral postnatal torsion (Gong et al, 1996). An enlarged contralateral testis (Huff et al, 1992) and absence of palpable intrascrotal “appendage” tissue (processus vaginalis, wolffian structures, or gubernaculum) are highly predictive of vanishing testis (Mesrobian et al, 2002). However, diagnosis of a vanishing testis requires documentation of blind-ending spermatic vessels in the abdomen, inguinal canal, or scrotum. Endocrine evaluation in cases of suspected bilateral vanishing testis (anorchia) include elevated basal serum gonadotropin levels and no response to hCG stimulation; however, gonadotropins may be unexpectedly low in mid childhood in boys who are also unresponsive to hCG (Lustig et al, 1987; Lee, 2000). Therefore laparoscopic or surgical documentation of anorchia is critical to avoid leaving small or dysgenetic abdominal testes in situ. AMH and inhibin-B levels may be useful in documenting the presence of functioning testicular tissue (Grumbach, 2005).
Inguinoscrotal ultrasonography and magnetic resonance imaging (MRI) are not usually helpful in the diagnosis and management of nonpalpable testis (Elder, 2002). However, if a testis is nonpalpable when evaluated by an experienced examiner, the sensitivity of ultrasonography in identifying inguinal testes is reported to be as high as 95% to 97%, and abdominal testes are also seen in some cases (Cain et al, 1996; Nijs et al, 2007). Some of these testes are “peeping,” however, and best mobilized via a laparoscopic approach. Similarly, although MRI can be useful in some cases to identify nonpalpable abdominal testes, its accuracy is variable and the procedure requires sedation in younger children and often may not change the management approach (Yeung et al, 1999; Siemer et al, 2000). The superior accuracy of magnetic resonance angiography in localizing and differentiating viable from vanished testes (96% of 23 nonpalpable testes in 21 boys, mean age 2.5 years) has not been replicated in a recent study of younger boys (57% of 29 testes in 26 boys, mean age 13 months) (Yeung et al, 1999; Desireddi et al, 2008). One indication for MRI may be identification of an ectopic abdominal testis not localized by laparoscopy.
Diagnostic laparoscopy followed by laparoscopic orchidopexy if the testis is abdominal has become the preferred approach of many clinicians. Important observations include size and position of the spermatic vessels and vas; testicular size, quality, and position; and patency of the internal inguinal ring. The combination of a closed internal ring and a blind-ending spermatic artery and vas is most consistent with an abdominal vanishing testis, whereas a hernia is frequently but not always associated with a viable abdominal or distal testis (Elder, 1994; Moore et al, 1994). An atretic spermatic cord coursing through a closed inguinal ring is suggestive of a distal vanishing testis, but this finding may be subjective; and, conversely, normal-appearing vessels may be associated with both viable and vanishing testes (Zaccara et al, 2004). Moreover, the laparoscopic view may suggest abdominal blind-ending vessels despite a testis being present distally or in an ectopic abdominal position (Zaccara et al, 2004; Kim et al, 2005; Ellsworth and Cheuck, 2009).
The need for excision and contralateral scrotal orchidopexy in cases of vanishing testis remains controversial. Germ cells and/or tubules are consistently present in 5% to 15% of excised testicular remnants (Moore et al, 1994; Tennenbaum et al, 1994; Turek et al, 1994; Cortes et al, 1995a; De Luna et al, 2003; Renzulli et al, 2005), but the risk of malignancy is unknown. A single case of carcinoma in situ was reported by Rozanski and associates (1996) in a testicular remnant. Excision may be appropriate for remnants distal to an internal ring to allow confirmation that a viable testis is absent, because the laparoscopic appearance of the spermatic vessels and processus vaginalis may be deceiving and may not reliably exclude the presence of an inguinal testis (Ellsworth and Cheuck, 2009). Storm and colleagues (2007) performed laparoscopic excision of vanishing testes in 56 patients without significant morbidity, although the position of the remnants in these patients was not clear. Because the testicular remnant is often in or near the scrotum, some advocate a primary scrotal approach when contralateral testicular hypertrophy (testicular length ≥ 1.8 cm) and a palpable scrotal “nubbin” are present (Belman and Rushton, 2003; Snodgrass et al, 2007). However, in cases of abdominal vanishing testes, a transscrotal search is time consuming and unproductive. Moreover, in cases of testicular-epididymal dissociation, scrotal nubbins may be mistaken for testicular remnants that would otherwise be identified by laparoscopy (Wolffenbuttel et al, 2000; De Luna et al, 2003). Laparoscopy is the procedure of choice to confirm or exclude the presence of a viable or remnant abdominal testis, unless a prominent scrotal nubbin is palpable with other clinical signs of monorchism.
The need for contralateral fixation of a solitary testis in cases of monorchism is not clear. The possibility that prenatal torsion is the etiology of vanishing testis (Gong et al, 1996) does not imply that the contralateral testis is likely to undergo a similar fate after the postnatal period. However, some surgeons recommend contralateral fixation to eliminate the risk of such a devastating complication (Rozanski et al, 1996) and/or because a contralateral bell-clapper deformity (incomplete testicular fixation to the tunica vaginalis) may be present (Bellinger, 1985; Al-Zahem and Shun, 2006).
Associated Pathology
Testicular Maldevelopment
Many observational studies of the histologic development of prepubertal cryptorchid testes have been published. More than 40 years ago, Mancini and associates (1965) systematically reported germ cell counts and related arrested development of spermatogonia with progressive loss in cryptorchid testes. Subsequently, several large series, some with additional normal autopsy (Hedinger, 1982) or affected (hernia, hydrocele) (Hadziselimovic et al, 1987a) control data and others that refer to these established age-dependent norms (Schindler et al, 1987; Huff et al, 1993; McAleer et al, 1995; Gracia et al, 2000; Cortes et al, 2001), have provided mainly consistent findings in cryptorchid boys. These data show that the number of spermatogonia per tubule (S/T) is reduced after infancy and fails to increase normally with age in cryptorchid and, to a lesser degree, in contralateral scrotal testes. The frequency of abnormal histology in the contralateral testis varies between studies, ranging from 22% to 95%, and is likely reflective of differences in patient populations, use of control data, and methodology. Moreover, variability within and between biopsy specimens from single testes is reported (Hedinger, 1982; Schindler et al, 1987). However, these data provide strong evidence that abnormal germ cell development is often present after early infancy in cryptorchid testes. The degree of pathology was similar in true ectopic, superficial inguinal pouch, and ascending testes (Herzog et al, 1992; Hutcheson et al, 2000b; Rusnack et al, 2002) and was more severe in limited samples from patients with myelomeningocele, posterior urethral valves, and prune-belly syndrome (Orvis et al, 1988; Patel et al, 2008). Findings are similar in retracted testes after hernia repair, suggesting primary instead of secondary cryptorchidism (Fenig et al, 2001). In other studies, higher S/T counts were correlated with reduced age-dependent interstitial fibrosis (Suskind et al, 2008), lower age at surgery, and increased likelihood of palpability (Tasian et al, 2009), but not volume (Noh et al, 2000), of cryptorchid testes. Regional differences in S/T counts were recently reported, potentially reflecting population differences in environmental exposures (Zivkovic et al, 2009).
Detailed studies of peritubular myoid and Sertoli cells in cryptorchidism are lacking, but their abnormal development or function may contribute to the observed germ cell abnormalities. The available data suggest disruption of prepubertal Sertoli cell morphology, failure of maturation at puberty, and evidence for reduced number after 4 months of age in cryptorchid testes (Lackgren and Ploen, 1984; Rune et al, 1992; Regadera et al, 2001; Zivkovic and Hadziselimovic, 2009). Reduced expression of type IV collagen, a product of both Sertoli and myoid cells that may function in cell-cell communication, was reduced in basement membranes of undescended and contralaterally descended testes (Santamaria et al, 1990).
Impaired transformation of gonocytes to spermatogonia is reported in cryptorchid testes and may be a key determinant of fertility potential. In normal testes, germ cell number decreases after birth as gonocytes either degenerate by apoptosis or migrate to the basement membrane and differentiate into spermatogonia (Hadziselimovic et al, 1986; Huff et al, 2001). Although the ratio of gonocytes to spermatogonia appears to be normal in cryptorchid testes at about 1.5 months of age, delayed disappearance of gonocytes and appearance of adult dark (Ad) spermatogonia occurs in the undescended as compared with the contralateral descended testis. Ad spermatogonia are likely the reserve stem cells of the germ cell pool (Dym et al, 2009). The mechanisms involved in migration and/or differentiation of gonocytes into stem cells are not clearly defined but appear to involve ADAM-integrin-tetraspanin complexes, retinoic acid, platelet-derived growth factor receptor, and c-KIT (Culty, 2009). Hadziselimovic and colleagues hypothesize that failure of gonocyte differentiation in cryptorchidism reflects failure of postnatal activation of the HPG axis (Hadziselimovic et al, 1986); however, although they occur concurrently there are no data confirming a direct relationship between the two events.
Kollin and colleagues (2006, 2007)
Related posts:
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Urodynamic and Video-Urodynamic Evaluation of the Lower Urinary Tract
Urodynamic and Video-Urodynamic Evaluation of the Lower Urinary Tract
 Neuropathic Dysfunction of the Lower Urinary Tract
Neuropathic Dysfunction of the Lower Urinary Tract
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


