- The rapid growth of pediatric hepatology as a specific and focused field of interest is attributable to the importance of the dramatic physiologic variables occurring in the maturing liver, as well as recognition of the unique nature of the liver diseases that affect infants and children.
- As with adults, the assessment of liver disease in children requires a careful history and physical examination; however, further investigations are directed by likely diagnoses, which differ significantly by age. Infants and young children, in particular, require careful assessment for congenital and inherited metabolic diseases.
- The assessment of liver disease in children involves directed laboratory investigations, radiologic investigations, and often a liver biopsy. The interpretation of these data requires the input of pediatric subspecialists.
Introduction
As in adults, the adequate evaluation of an infant, child, or adolescent with suspected liver disease involves an appropriate and accurate history, a carefully performed physical examination, and skillful interpretation of signs and symptoms. Further evaluation is aided by judicious selection of diagnostic tests, and in pediatrics special consideration is given to the selection and timing of invasive investigations. The initial approach for the health-care provider first encountering a child with suspected liver disease may address some of the following issues, including: (1) is liver disease present and, if so, what is the nature of the liver disease; (2) what is the severity; and (3) is there specific treatment available. Subsequent considerations may include: (4) how to monitor the response to treatment; and (5) the prognosis of the liver disease.
Clinical Presentations and Common Diagnoses
Asymptomatic Elevation of Liver Transaminase Levels
With routine laboratory testing now automated and so easily available, physicians often encounter the child with asymptomatic elevation of serum liver transaminase levels. Serum aminotransferase levels are sensitive indicators of liver cell injury and are helpful in recognizing hepatocellular diseases such as those caused by viral, drug-induced or autoimmune hepatitis, non-alcoholic fatty liver disease, or certain chronic liver diseases associated with metabolic conditions previously undiagnosed in the child. It is noteworthy to mention that the normal range for serum aminotransferase levels varies widely among laboratories, along with the recent realization that upper limits of normal may be too high for pediatric patients. This may result in missed opportunities to diagnose liver disease in children. If the results of repeated tests are abnormal, further evaluation is indicated to assess potential etiologic conditions (Box 3.1). A reliable body of literature to enable unequivocal recommendations for the diagnostic evaluation of children with abnormal liver chemistry tests is lacking.
- Chronic viral hepatitis (hepatitis B, hepatitis C, hepatitis D)
- Medication (drug-induced hepatitis)
- Non-alcoholic fatty liver disease (NAFLD)
- Autoimmune hepatitis
- Alcohol
- Other autoimmune disorders such as systemic lupus erythematosus
- Metabolic disorders associated with chronic liver disease (in order of prevalence)
- α1-antitrypsin deficiency
- Wilson disease
- Tyrosinemia
- Glycogen storage disease
- Galactosemia
- α1-antitrypsin deficiency
- Celiac disease
- Inherited disorders of muscle metabolism
- Acquired muscle diseases
- Cardiomyopathy (serum AST levels
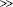 serum ALT levels)
serum ALT levels)
- Cystic fibrosis
- Other rare metabolic conditions: Niemann–Pick disease
Jaundice
Jaundice, when it affects children, most commonly presents in the first few months of life. The most common cause of jaundice in the neonate is physiologic and is manifested by predominantly unconjugated hyperbilirubinemia. Any infant jaundiced beyond 14 days of life warrants a fractionated serum bilirubin. Conjugated hyperbilirubinemia (>20% of total bilirubin) is always pathologic and requires assessment by a pediatric hepatologist. A discussion of unconjugated hyperbilirubinemia is beyond the scope of discussion here. Generally, in the assessment of conjugated hyperbilirubinemia, historical clues are unhelpful in the diagnosis, except to elucidate if the infant has recently been systemically unwell. There are a multitude of diagnoses that can cause jaundice before the age of 1 year, which typically present with conjugated hyperbilirubinemia (Box 3.2). Some of these diseases require a prompt diagnosis, for example biliary atresia, treatable metabolic disease, and it is therefore important to rule out these diagnoses without delay. In particular, it is optimal for a baby with biliary atresia to undergo corrective surgery before 60 days of age to maximize the likelihood of successful long-term survival with native liver. Infants with biliary atresia or a choledochal cyst may be jaundiced but look quite well clinically. Stool color is a key part of the history as both of these diagnoses lead to obstructive jaundice, which can cause a pale or acholic stool. Infants with Alagille syndrome may have evidence of cardiac involvement in association with jaundice. Infants with the metabolic diseases tyrosinemia, galactosemia, or hypopituitarism are likely to present early with jaundice and will often look unwell—there is a classic association of Escherichia coli sepsis in association with galactosemia. However, any bacterial infection can manifest with jaundice in the neonatal period. Urinary tract infections commonly present within the first 2 months of life with jaundice, lethargy, or poor feeding but rarely with fever. Children with progressive familial intrahepatic cholestasis (PFIC) present later (under 6 months) and have severe pruritus that is disproportionate to the degree of jaundice. The child will also often have evidence of failure to thrive.
- Biliary atresia
- Choledochal cyst
- Alagille syndrome
- α1-antitrypsin deficiency
- Progressive intrahepatic cholestasis (PFIC)
- Tyrosinemia
- Galactosemia
- Hypopituitarism/hypothyroidism
- Bacterial sepsis, particularly urosepsis
- Viral infections: cytomegalovirus, enterovirus
- Congenital infections: syphilis, rubella
- Hepatitis A in day care centers
- Hepatitis A infection
- Autoimmune hepatitis
- Primary sclerosing cholangitis
- Choledochal cyst
- Obstructive gallstones (common bile duct stones, i.e. may never have been in gall bladder e.g. MDR3 mutation)
- Wilson disease
- Benign recurrent intrahepatic cholestasis (BRIC)
- Drug-induced: estrogens, erythromycin, acetaminophen
Older children presenting with jaundice generally appear well. Children with hepatitis A infection may have a history of travel abroad or eating contaminated food. Children with obstructing gallstones may have a history of acute recurrent abdominal pain. Obstructing stones or a choledochal cyst often manifest with the classic triad of jaundice, pale stools, and dark urine. Primary sclerosing cholangitis and autoimmune hepatitis may present with non-specific symptoms of malaise and fatigue. There may be a positive family history of other autoimmune diseases. Children with inflammatory bowel disease in association with primary sclerosing cholangitis may report, when asked, altered bowel habits. A child with Wilson disease may have a history of neurologic symptoms or deterioration in school performance. Finally, patients with benign recurrent intrahepatic cholestasis (BRIC) reveal a history of intermittent jaundice, often with very severe pruritus. These episodes are followed by asymptomatic intervals and they never develop progressive liver disease.
Hepatomegaly, Splenomegaly, and Hepatosplenomegaly
Enlargement of the liver and/or spleen commonly occurs with many of the aforementioned causes of jaundice, as well as with any disease causing portal hypertension. This section will focus on etiologic conditions in which organomegaly rather than jaundice is the primary presenting feature. This is a large group of conditions, which may be primarily hepatic or extrahepatic (Box 3.3).
- Storage disorders
- Glycogen storage diseases
- Lysosomal disorders: Gaucher disease, Niemann–Pick C
- Glycogen storage diseases
- Portal vein thrombosis with cavernous transformation
- Metabolic liver diseases: α1-antitrypsin deficiency
- Cirrhosis of any cause, e.g. fatty liver disease
- Congenital hepatic fibrosis
- Infection: chronic viral hepatitis, EBV infection
- Malignancy
- Acute lymphoblastic leukemia
- Hepatoblastoma
- Acute lymphoblastic leukemia
- Rheumatologic diseases, e.g. juvenile rheumatoid arthritis
- Hematologic disorders, e.g. sickle cell disease, hereditary spherocytosis
Storage disorders are important causes of pediatric liver disease. Glycogen storage diseases usually present with hepatomegaly and hypoglycemia. Gaucher disease is the most common lysosomal storage disease; the majority of cases are discovered because of hepatomegaly. Associated neurologic findings are only seen in a minority of cases. Niemann–Pick C is a disorder of lipid trafficking and is usually associated with symptomatic, progressive neurocognitive dysfunction such as clumsiness and poor school performance. Physical examination reveals splenomegaly and/or hepatomegaly in most instances.
Cavernous transformation of the portal vein following thrombosis is a relatively common cause of splenomegaly, with or without an initial presentation of gastrointestinal bleeding. This is described in more detail in Chapter 5. Congenital hepatic fibrosis is a fibrocystic disease of the liver, often related to autosomal recessive kidney disease but it can also be associated with other syndromes. This condition may also present with splenomegaly and/or gastrointestinal bleeding.
Fulminant Liver Failure
All health-care providers must be aware that children in acute liver failure may not fulfill the expected altered mental status criteria of hepatic encephalopathy required for the classic definition of fulminant liver failure in adults. In a pediatric patient with no known liver disease, who has an uncorrectable coagulopathy with clinical and/or biochemical evidence of acute liver injury, prompt admission is critical. Immediate initiation of appropriate supportive therapy focused on preventing or treating complications, and arranging for early referral to a pediatric transplant center, may be life saving.
A number of difficulties exist with applying the classic definition of “fulminant liver failure” in pediatric patients. Herein, “pediatric acute liver failure (PALF)” will be used interchangeably with fulminant liver failure. Firstly, hepatic encephalopathy is often difficult to recognize and diagnose in infants and small children, particularly in the early stages of illness, such that PALF may be present without clinical evidence of encephalopathy. Secondly, the presence of uncorrectable coagulopathy is an important (overt bruising may occur), consistent and reliable finding in children with PALF, even in the absence of hepatic encephalopathy. Thirdly, PALF can present in infants less than 8 weeks of age, and even during the first days of postnatal life. For these and other reasons, a consensus working definition for the clinical condition of acute liver failure in children was derived and currently being applied by the NIH funded Pediatric Acute Liver Failure (PALF) Study Group:
and
Related posts:
 16 Drug-induced liver disease
16 Drug-induced liver disease
 11 Management of the complications of liver transplantation
11 Management of the complications of liver transplantation
 20 Chronic cholestatic liver disease and its management in adults and children
20 Chronic cholestatic liver disease and its management in adults and children
 12 Acute viral hepatitis in adults and children: hepatitis A, B, C, D, E, and others
12 Acute viral hepatitis in adults and children: hepatitis A, B, C, D, E, and others
 26 An internist’s approach to radiologic examination of the liver
26 An internist’s approach to radiologic examination of the liver
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


