Alcohol distribution is dependent on blood flow, with vascular organs such as the brain rapidly equilibrating with plasma levels. Alcohol is poorly soluble in lipids, possibly explaining the higher plasma concentrations found in females compared to males that have consumed the same amount of ethanol.
Alcohol cannot be stored and obligatory oxidation must take place, predominantly in the liver. The healthy individual cannot metabolize more than 160–180 g/day. Alcohol induces enzymes used in its catabolism, and the hazardous drinker, at least while the liver is relatively unaffected, can metabolize more.
Alcohol to Acetaldehyde
Between 80 and 85% of ethanol oxidation is by initial conversion to acetaldehyde catabolized by alcohol dehydrogenase (ADH) (Fig. 25.2). This takes place in the cytosol. The resulting increase in the ratio of NADH/NAD, which is further increased by acetaldehyde oxidation, is partly responsible for the metabolic imbalances that occur following alcohol ingestion and has been considered to play a major role in the initial pathogenesis of alcohol-induced fatty liver.
Fig. 25.2. The three pathways of alcohol oxidation. MEOS, microsomal ethanol oxidizing system; CYP2E1, cytochrome P4502E1.
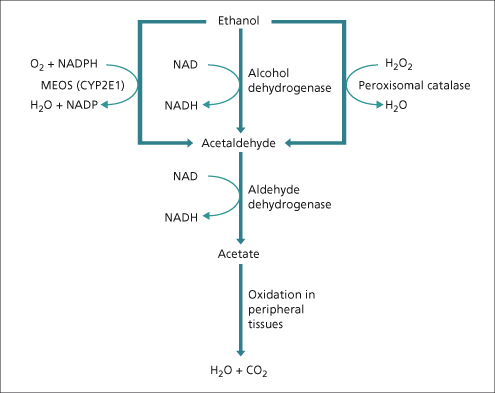
Most of the remaining ethanol is metabolized by the microsomal ethanol-oxidizing system (MEOS) pathway, an accessory pathway that principally involves a specific alcohol-inducible form of cytochrome P450, designated CYP2E1 [2]. The induction of CYP2E1 in hazardous drinkers may explain their increased susceptibility to hepatotoxicity by other drugs that are converted to toxic metabolites by this enzyme system. An important example of this phenomenon is the increased susceptibility to the toxic effects of paracetamol (acetaminophen), where severe liver damage has been reported in dependent drinkers taking therapeutic doses [3].
Acetaldehyde to Acetate
Most of the acetaldehyde formed from ethanol oxidation is further oxidized in the liver to acetate by aldehyde dehydrogenases (ALDHs). Acetate may be oxidized to carbon dioxide and water, or converted by the citric acid cycle to other compounds, including fatty acids. The inactive form of ALDH (ALDH2*2) is present in about 50% of Orientals. The accumulation of acetaldehyde may account for the flushing seen with alcohol consumption in homozygotes.
Pathogenesis
Rodents given alcohol develop only a fatty liver. However, they cannot match the consumption achieved by humans, who may take 50% of total calories as alcohol. This level can be achieved in the baboon, which develops cirrhosis after 2–5 years of high alcohol consumption. Evidence for a direct hepatotoxic effect of alcohol, as opposed to an indirect effect related to the associated nutritional changes, comes from studies in human volunteers who, after 300–600 mL (10–20 oz) of 86% proof alcohol daily for 8–10 days, develop fatty change and electron microscopic abnormalities on liver biopsy [4].
Some of the pathways through which alcohol may cause liver injury are described below. Unfortunately, this improved knowledge of disease mechanisms has, to date, not led to a significant improvement in the therapy of alcoholic liver disease.
Pathogenesis of Steatosis
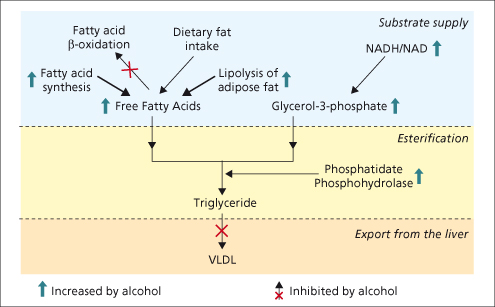
The accumulation of triacylglycerol (TAG) within the liver is an early and reversible effect of alcohol consumption. Alcohol increases peripheral lipolysis and the altered liver redox potential increases fatty acid synthesis. This increase in substrate supply (glycerol and free fatty acids) enhances the rate of esterification, resulting in TAG accumulation. This is compounded by the alcohol-induced inhibition of the enzyme which controls TAG export from the liver, microsomal triglyceride transfer protein (MTP) [5] (Fig. 25.3).
Fig. 25.3. The multiple mechanisms through which ethanol metabolism can lead to fatty liver. Ethanol causes fatty liver by increasing substrate supply, increasing fat esterification to triglyceride and reducing the export of very low density lipoprotein (VLDL) from the liver.
Oxidative Stress and Lipid Peroxidation
In alcohol-related liver disease (ALD) the generation of pro-oxidants overwhelms the endogenous antioxidant systems, resulting in lipid peroxidation. These pro-oxidants can come directly from ethanol metabolism or from activated phagocytes. Liver injury is compounded by the depletion of endogenous cellular, particularly mitochondrial, antioxidants in hazardous drinkers.
Acetaldehyde
Acetaldehyde is generated by both ADH and the MEOS systems and may account for many of the features of acute alcoholic hepatitis (Table 25.1). Acetaldehyde is extremely reactive and toxic; it binds to phospholipids, amino acid residues and sulphydryl groups. It can produce altered surface antigens and depolymerize proteins, altering their folding. When abnormally folded or unfolded proteins build up in the endoplasmic reticulum (ER), this results in a phenomenon known as ‘ER stress’ [6]. ER stress induces further lipid synthesis, antioxidant depletion and ultimately apoptosis.
Table 25.1. Possible hepatotoxic effects of acetaldehyde
| Induction of steatosis through altering the redox potential |
| Increasing sensitization to TNF-α mediated hepatocyte |
| necrosis |
| Binding to host proteins: |
| affecting folding and inducing endoplasmic reticular stress |
| affecting function (e.g. microtubules) |
| forming neoantigens |
Endotoxin and Cytokines
A complex relationship exists between endotoxin, Kupffer cell activation and the release of cytokines and chemokines. Endotoxin is increased in the blood of hazardous drinkers [7]. This is related to increased intestinal bacterial flora, increased gut permeability and reduced endotoxin scavenging by the reticuloendothelial system (Fig. 25.4). Endotoxin results in the release of cytokines and reactive oxygen species from Kupffer cells. In alcoholic hepatitis, tumour necrosis factor-α (TNF-α) and interleukin-8 (IL-8) production are particularly increased [8,9].
Fig. 25.4. Alcohol, endotoxinaemia and cytokine production. RE, reticuloendothelial; TNF, tumour necrosis factor; IL, interleukin.
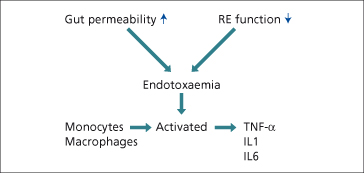
The biological effects of certain cytokines resemble the clinical and histological manifestations of ALD (Table 25.2). TNF-α can induce steatosis, the production of reactive oxygen species (ROS) and hepatocyte apoptosis. IL-8 is involved in the recruitment and activation of neutrophils.
Table 25.2. Biological effects of cytokines and clinical features of alcohol-related liver disease
| Change | Alcohol-related liver disease | Cytokines |
| Fever | + | + |
| Anorexia | + | + |
| Muscle wasting | + | + |
| Hypermetabolism | + | + |
| Neutrophilia | + | + |
| Decreased albumin | + | + |
| Collagen disposition | + | + |
| Increased triglycerides | + | + |
| Decreased bile flow | + | + |
| Shock | + | + |
Immunological Liver Damage
Protein adducts formed from ethanol metabolites and host proteins can act as neoantigens to incite humoral B-cell and cytotoxic T-cell lymphocyte responses in ALD. Antibodies can be shown against acetaldehyde protein adduct-derived epitopes [10] and hydroxyethyl radical–CYP2E1 adducts [11]. Antibodies can also be seen to native CYP2E1, suggesting that autoimmune mechanisms may play a role in alcohol-related liver disease [12]. The true importance of immunological mechanisms is not clear as they may represent an epiphenomenon whereby immune responses are generated to proteins released from hepatocytes damaged through other mechanisms.
Alcohol and Fibrosis
The proliferation and activation of stellate cells in ALD is promoted by Kupffer cells and hepatocytes. Kupffer cells induce collagen synthesis through the production of transforming growth factor-β (TGF-β), TNF-α and ROS. Hepatocytes induce fibrosis through the production of ROS or through apoptosis. TGF-β is produced when apoptotic hepatocytes are phagocytosed and this in turn can activate stellate cells [13].
Alcohol and Cancer
Alcohol consumption is associated with hepatocellular carcinoma and several extrahepatic cancers (Table 25.3). The mechanisms are likely to be related to lipid peroxidation and DNA mutagenesis, reduced DNA methylation and immunosuppression.
Table 25.3. Alcohol and cancer
| Mouth |
| Pharynx |
| Larynx |
| Oesophagus |
| Colon |
| Breast |
| Liver |
Susceptibility
Environmental Factors
Dose of Alcohol
The average intake of alcohol in a large group of male dependent cirrhotic patients was 160 g/day for 8 years. The risk of developing ALD begins at 30 g/day of ethanol [14] but for most individuals the dose that confers a significant risk is greater than 80 g alcohol daily. The duration of consumption is also important. In one study, neither cirrhosis nor alcoholic hepatitis were seen in patients who consumed an average of 160 g of ethanol per day for less than 5 years, whereas 50% of patients consuming high levels of alcohol for an average of 21 years developed cirrhosis [15]. Liver injury appears to be unrelated to the type of beverage; reports that wine drinkers have a lower risk than beer and spirit drinkers and that drinkers of mixed beverages have a higher risk than those keeping to a single type of drink are probably explained by confounding lifestyle factors associated with particular drinking patterns. Continued daily imbibing is more dangerous than intermittent consumption when the liver is given the opportunity to recover; it is therefore recommended that individuals should have at least two alcohol-free days per week.
ALD and dependence do not necessarily go together. Those who develop alcohol-related liver damage are often not dependent on alcohol. Most dependent patients have normal liver function [16].
Diet
Cirrhosis mortality has been linked with diets high in pork (high in linoleic acid) consumption and unsaturated fats [17] and low in carbohydrate [18]. Obesity and associated hyperglycaemia increase the incidence of all stages of ALD in heavy drinkers [19,20].
Genetic Factors
Gender
Hazardous drinking is increasing among women owing to a decline in the social stigma and the increased availability of alcohol. Women are less likely to be suspected of alcohol abuse; they present at a later stage, are more susceptible to liver injury and are more likely to relapse after treatment [21]. This may be related to the reduced volume of distribution of alcohol, or the fact that, in animal models at least, oestrogen increases gut permeability to endotoxin [22]. Women are more likely to progress from alcoholic hepatitis to cirrhosis even if they abstain [23].
Non-Gender-Linked Genetic Factors
Patterns of alcohol drinking are, at least partially, inherited; however, no specific genetic variants have been reproducibly associated with susceptibility in large studies. Susceptibility to liver disease may also have an inherited component. Concordance rates for alcohol-related cirrhosis are three times higher in monozygotic than in dizygotic twin pairs [24]. Alcohol-related liver damage is a polygenic disorder so multiple polymorphisms are likely to contribute. They are likely to be in genes controlling fat accumulation, oxidative stress, endotoxin-mediated release of proinflammatory cytokines and immunological damage [25].
Histological Features
Fatty Liver
Fat (steatosis) accumulates predominantly in zones 3 and 2 although in the more severely affected, the fatty change is diffuse. Typically the fat is in a macrovesicular (large droplet) form although it can also be in a microvesicular (small droplet) form.
Large fat droplets appear in hepatocytes within days of excess alcohol ingestion. Microvesicular fat probably reflects the presence of mitochondrial injury and resulting inhibition of fatty acid oxidation. In support, hepatic mitochondrial DNA deletion has been reported in patients with alcohol-related fatty liver. Fatty change can be quantified according to the proportion of hepatocytes that contain fat.
Alcoholic Hepatitis
The full histological picture of a florid, acute alcoholic hepatitis is relatively rare. Typical features include some or all of the following:
Ballooning Degeneration.
Hepatocytes are swollen with granular cytoplasm often dispersed into fine strands. The nucleus is small and hyperchromatic. The ballooning is due to retention of water and to failure of the microtubular excretion of protein from the hepatocyte.
Acidophilic Bodies.
These represent hepatocyte apoptosis.
Mallory–Denk Bodies.
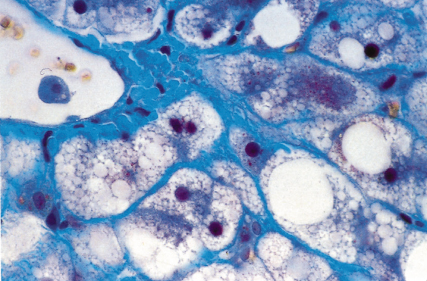
These are seen on haematoxylin and eosin stained sections as purplish-red intracytoplasmic inclusions. They may be more obvious with Masson’s trichrome or chromophobe aniline blue stains. They consist of clumped organelles—largely intermediate filaments—and may target the hepatocyte for destruction. The Mallory-containing cell is surrounded by a satellite of polymorphs (Figs 25.5, 25.6).
Fig. 25.5. Acute alcoholic hepatitis with ballooning degeneration, Mallory-Denk bodies and satellitosis (neutrophil polymorph infiltrate around hepatocytes). (H & E × 120). Photo provided courtesy of A.D. Burt.
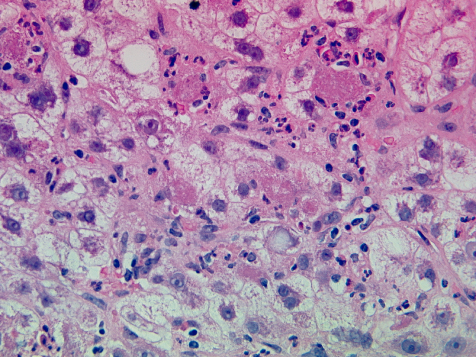
Fig. 25.6. Acute alcoholic hepatitis. Hepatocytes are ballooned and contain micro- and macrovesicular fat and clumps of purplish-red Mallory’s alcoholic hyaline. (Chromophobe aniline blue, ×100.)
Giant Mitochondria.
These form globular intracytoplasmic inclusions seen by light microscopy using a Masson trichrome stain.
Fibrosis.
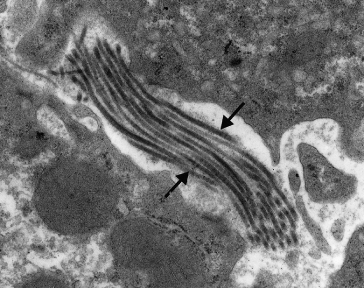
Collagen deposition is usually maximal in zone 3. The fibres are perisinusoidal and enclose normal or ballooned hepatocytes. The pericellular fibrosis is like lattice or chicken wire and has been termed ‘creeping collagenosis’ (Fig. 25.7) [26]. Collagenization of the space of Disse is shown by electron microscopy (Fig. 25.8) and is associated with a reduction in the porosity of the sinusoidal lining [27]. These changes interfere with the exchange of substances between plasma and the hepatocyte cell membrane and contribute to portal hypertension. Associated lesions in terminal and sublobular veins include lymphocytic phlebitis, gradual obliteration and eventual veno-occlusion [28].
Fig. 25.7. Advanced zone 3 collagenosis with fatty change. A thickened hepatic vein can be seen bottom right. (Chromophobe aniline blue, ×100.)
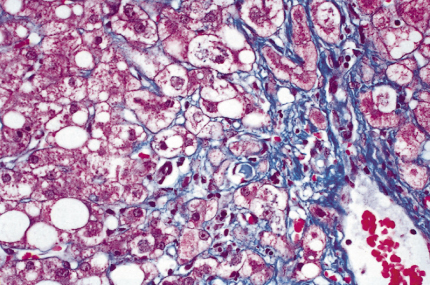
Fig. 25.8. Electron micrograph of liver in a patient with alcoholic liver disease. Note the deposition of collagen fibrils in Disse’s space (arrowed). This could interfere with oxygen and metabolite exchange between blood and hepatocytes.
Portal Zone.
Changes in the portal zone are inconspicuous and mild chronic inflammation is seen only in the advanced case. Zone 1 fibrosis if present is not now thought due to previous pancreatitis (Fig. 25.10) [29].
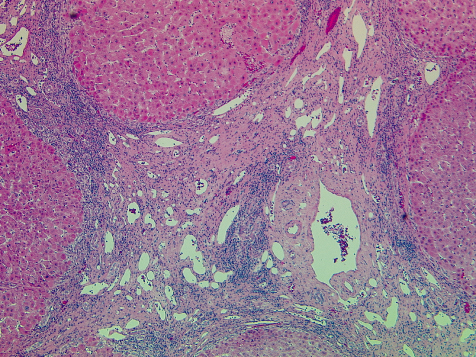
Fig. 25.9. End stage cirrhotic alcoholic liver disease. Established micronodular cirrhosis with areas of parenchymal extinction. (H & E, ×120.) Photo courtesy of A.D. Burt.
Cholestasis.
Cholestasis in bile canaliculi is a feature of all types of alcohol-related liver disease. It is strongly associated with decreased survival.
These histological patterns form a spectrum from minimal hepatitis to an advanced, probably irreversible, picture, where necrosis is more extensive and fibrosis is prominent. Alcohol-related hepatitis is a precursor of cirrhosis and in the majority of cases is superimposed on established cirrhosis.
Cirrhosis
Classical alcohol-related cirrhosis is micronodular (Fig. 25.9
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


