ERM, electronic medical records; DILI, drug-induced liver injury; ICD, International Classification of Diseases; NR, not reported.
Data shown in Table 24.2 were accumulated over several years by evaluating between 732 and 1 636 792 individuals. The prevalence of hepatotoxicity, reported in five studies, ranged between 0.7 and 22.8%, although the latter percentage was an outlier. Estimated incidences, reported in three studies, were 1/100 000, 2.4/100 000 and 40.6/100 000. In one study, paracetamol (acetaminophen) hepatotoxicity exceeded the true idiosyncratic liver injury cases by a factor of 5 [29]. Another study focused on identifying specific drugs that cause liver injury (amoxicillin clavulanate, phenytoin, valproate and isoniazid) [32].
Prospective Cohort and Registry Studies.
Prospective studies involve gathering consecutive cases of drug-induced liver injury either at a single centre or through multicenter registries. One representative prospective study, performed at a Swedish University out-patient clinic, reported that 77 (6.6%) of 1164 successive patients with liver disease had possible drug-induced liver disease [34].
Three registry studies are shown in Table 24.3; two collect all cases of idiosyncratic drug-induced liver injury [35,36] while one accepts only cases of fulminant hepatic failure [37,38]. The Spanish Registry has been assembling cases over a 10-year period from 32 referral centres [35]. Among 570 cases submitted, 461 were considered bona fide instances of drug-induced liver injury; 49% were female, 71% were jaundiced, 51% were hospitalized and 7% died or required liver transplantation. The annual incidence of hepatotoxicity was 34.2 ± 10.7 cases per 100 000 inhabitants, and the annual incidence of severe drug-induced liver injury was 16.6 ± 6.7 cases per 100 000 inhabitants.
Table 24.3. Prospective registry studies of patients with drug-induced liver injury
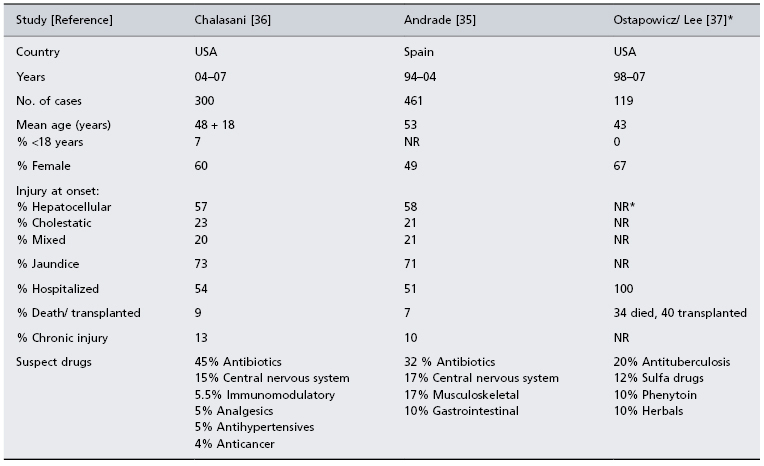
* All patients had acute liver failure at entry with INR >1.5 and encephalopathy. The median alanine aminotransferase at presentation was 571 IU/mL.
A second registry is part of the ongoing US Drug-Induced Liver Injury Network (DILIN) study, which recently reported findings in the first 300 consecutive cases collected [36]. The mean age of enrollees was 48 years, 60% were female and 7% were children. The median latency to liver injury was 42 days, and 54% were hospitalized. A single prescription drug was implicated in 73% of cases, 9% were attributed to herbals and dietary supplements and 18% had received more than one suspect medication. The most commonly implicated single drugs received were antimicrobials (45.5%), neuropsychiatric medications (15%) and immunomodulatory agents (5.5%). Among the 9% of patients requiring transplantation or who died, fewer than one-half died of liver disease. Liver injury persisted beyond 6 months in 14% of the cases.
In the continuing US Acute Liver Failure Study Group registry, 40 (13%) of the first 308 reported cases from 23 referral centres consisted of idiosyncratic drug reactions, which included isoniazid, bromfenac and troglitazone [37]. The most commonly implicated drug, however, was paracetamol. In a follow-up report, 119 (11.5%) of 1033 consecutive cases of acute liver failure were attributed to idiosyncratic drug-related injury [38]. The mean age of patients was 43 years, 67% were female and only 26% survived without a transplant. The most commonly implicated medications in this follow-up report were antibiotics, which included antituberculosis drugs (20%), sulfa compounds (12%), phenytoin (10%) and herbals (10%).
Perhaps the most accurate data regarding the true incidence of hepatotoxicity come from a population-based prospective study conducted among 81 000 inhabitants of Northern France [16]. All potential cases of hepatotoxicity were referred to a central study team over a 3-year period. Ninety-five cases of hepatotoxicity were suspected, but only 34 were assessed as having probable hepatotoxicity. Their mean age was 50.9 years, 65% were female, and 6% died from liver injury. Only 30% had been referred to a gastroenterology specialist, indicating that the majority were managed by primary care physicians. Suspect medications included antibiotics (25%), psychotropics (22%), hypolipidaemics (12%) and non-steroidal anti-inflammatory drugs (10%). The annual incidence of drug-induced liver injury was estimated at 13.9 ± 2.4 episodes per 100 000 patient years and did not differ substantially by gender and age. This annual incidence of hepatotoxicity was over 16-fold higher than that reported to health authorities, signifying a high rate of under-reporting of drug-induced liver injury in the general population.
Asian Countries.
The epidemiology and risk factors for drug-induced liver injury have been less well studied in Asian than in Western countries. Available reports from Asia indicate, however, that implicated medications are more likely to be herbal and/or other traditional ‘medicines’ [39,40].
Expressions of Hepatotoxicity
There is virtually no form of liver disease, acute or chronic, that drug-induced liver injury cannot simulate (Table 24.4) [41–43]. It is this fact that confounds the adjudication process.
Table 24.4. Expressions of drug-induced liver injury
| Type of injury | Representative responsible drug(s) |
| Acute liver diseases | |
| Acute hepatocellular injury | Numerous drugs such as isoniazid, rifampicin, methyl dopa, telithromycin, ketoconazole, diclofanac |
| Mononucleosis-like | Sulfonamides, phenytoin, dapsone |
| Fulminant hepatitis | Paracetamol (acetaminophen) |
| Bland cholestasis | Anabolic/ androgenic steroids, ciclosporin |
| Cholestatic hepatitis | Chlorpromazine, erythromycin, amoxicillin–clavulanate, clarithromycin |
| Chronic liver diseases | |
| Chronic hepatitis | Methotrexate, lisinopril, trazodone, uracil |
| Autoimmune hepatitis | Nitrofurantoin, minocycline, methyldopa, oxyphenisatin |
| Macrovesicular hepatitis | Corticosteroids, methotrexate, asparaginase, alcohol, halothane |
| Microvesicular hepatitis | Valproic acid, tetracyclines, cocaine, amiodarone |
| Steatohepatitis | Amiodarone, griseofulvin, perhexillene maleate |
| Cirrhosis | Methotrexate, amiodarone |
| Granulomatous hepatitis | Allopurinol, rosiglitazone, sulfonamide, phenylbutazone, quinidine |
| Primary biliary cirrhosis-like | Chlorpromazine, erythromycin, amoxicillin–clavulanate, haloperidol |
| Peliosis hepatic | Anabolic steroids, oral contraceptives |
| Portal vein thrombosis | Oral contraceptives |
| Sinusoidal obstructive syndrome | Pyrrolozidine alkaloids, adriamycin, floxuridine, oncotherapy |
| Nodular transformation | Anabolic and contraceptive steroids |
| Adenoma | Anabolic and contraceptive steroids |
| Hepatocellular carcinoma | Thorotrast, anabolic and contraceptive steroids |
| Cholangiocarcinoma | Thorotrast |
| Angiosarcoma | Vinyl chloride, inorganic arsenicals |
Acute Liver Diseases (Table 24.4).
Drug-induced liver injury presents most commonly as acute liver disease, hepatocellular exceeding cholestatic injury. The injury may imitate acute viral hepatitis, including a mononucleosis-like picture, ranging from mild, asymptomatic illness to more severe injury, characterized by increased levels of both aminotransferases and serum bilirubin, and even fulminant hepatic failure. Hepatotoxicity may also simulate acute obstructive jaundice, presenting either as bland cholestasis with normal serum enzymes, or as cholestatic hepatitis, characterized by increases in both serum bilirubin and alkaline phosphatase. A third form of acute injury is macrovesicular and microvesicular steatosis.
Chronic Liver Diseases (Table 24.4).
Drugs can also cause the entire spectrum of chronic liver diseases with histology resembling chronic viral or autoimmune hepatitis, macro- or microvesicular steatosis or steatohepatitis, and even fully established cirrhosis. Hepatotoxicity can also simulate chronic cholestatic diseases, presenting as chronic granulomatous liver disease or as a picture resembling primary biliary cirrhosis or sclerosing cholangitis, occasionally culminating in the vanishing bile duct syndrome. Drugs can also cause such vascular disorders as peliosis hepatis, hepatic vein thrombosis, and the sinusoidal obstruction syndrome (previously veno-occlusive disease). Finally, drugs may induce hyperplasia such as nodular transformation, as well as benign (adenomas) and malignant (hepatocellular carcinoma, cholangiocarcinoma, angiosarcoma) neoplasms.
Classification of Hepatotoxicity
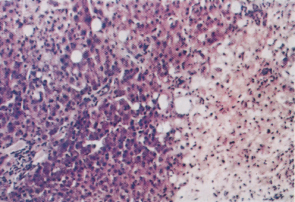
Direct hepatotoxins typically cause dose-dependent liver damage and zonal hepatocyte necrosis, an effect readily reproducible in animals. Examples are carbon tetrachloride and paracetamol which cause centrilobular necrosis, yellow phosphorus which causes midzonal injury, and allyl alcohol which causes periportal injury (Fig. 24.1) [44–46]. Few direct hepatotoxins are available for routine public use with the notable exception of paracetamol. A recent study actually demonstrated that almost one-third of healthy volunteers administered a therapeutic dose of 4 g of paracetamol per day developed mild elevations in serum alanine aminotransferase (ALT) values together with detectable serum paracetamol-protein adducts [47].
Fig. 24.1. Accidental carbon tetrachloride poisoning. To the right of the section, liver cells are necrotic and show hydropic degeneration and fatty change. Surviving cells to the left of the section show occasional fatty change. The portal zones are unaffected.
‘Idiosyncrasy’ derives from a Greek word that means ‘a mixture of one’s own self’ [13]. The term is applied to drugs that cause injury in a minority of individuals who appear uniquely susceptible due to alteration in host metabolism, uptake or processing of the drug. Unlike direct hepatotoxins, idiosyncratic hepatotoxicity does not present as a single type or pattern of liver injury. Generally, idiosyncratic liver injury is diffuse consisting of necrosis, cholestasis and/or steatosis that involves the entire lobule, accompanied by variable inflammatory cell infiltrates consisting of lymphocytes, plasma cells and eosinophils.
Idiosyncratic drug-induced liver injury may present with hypersensitivity or immunoallergic features in a minority of patients or as an autoimmune form of liver disease. The former is characterized by a generally short latency to liver injury onset with fever, rash, lymphadenopathy and arthralgias, together with eosinophilia and lymphocytosis [48]. Some drugs typically associated with this response include the sulfonamides, procainamide and the penicillins.
In some instances, circulating antibodies to liver and kidney endoplasmic reticulum (antiliver/ kidney microsomal antibodies, anti-LKM) may develop. These antibodies result from an acquired immune response to a new antigen created by covalent binding between a reactive metabolite and a hepatocellular protein. Anti-LKM antibodies frequently react with cytochrome P450s (CYPs) but may also react with other drug-metabolizing enzymes [49,50]. Antienzyme antibodies have been found in hepatotoxicity caused by tienilic acid, dihydralazine and halothane [51–53]. The current concept is that a highly reactive metabolite binds covalently to or damages the enzyme that produced it [54]. Antibodies form if this enzyme is antigenic and advances outside the hepatocyte where it becomes attached to antigen-presenting cells. Because antibodies recognize different proteins from liver injury due to different drugs (i.e. bioactivating enzymes differ depending upon drug structure), antiliver antibodies should be useful in some instances in assessing causality. Aromatic anticonvulsants have been associated with antibodies to CYP3A11 and halothane has been associated with antibodies to the E2 subunit of pyruvate dehydrogenase. Most medications that cause hepatotoxicity, however, are not associated with a characteristic drug-metabolizing autoantibody profile.
Some drugs, such as aldomet, minocycline and nitrofurantoin, can cause an autoimmune form of liver injury characterized by a liver biopsy displaying prominent plasma cell infiltration, the presence of autoantibodies such as the antinuclear antibody (ANA) and smooth muscle antibody (SMA), and variable increases in serum immunoglobulin levels [55–57]. This poses the problem of distinguishing drug-induced from a pre-existing but unrecognized idiopathic form of autoimmune hepatitis. One clue to exclude pre-existing autoimmune hepatitis is that the liver injury resolves and the autoantibodies disappear after discontinuing the drug, although, in some instances, short- or long-term treatment with corticosteroids is necessary, particularly in those with jaundice, to induce remission of drug-induced autoimmune hepatitis.
Predictors of Susceptibility and Outcome in Drug-Induced Liver Injury
Type of Presentation.
It has long been assumed that drug-induced hepatocellular injury, particularly if accompanied by jaundice, is more likely than cholestatic liver disease to have a fatal outcome. This assumption is not supported by recent data from the Spanish, Swedish, and US registry studies that have found that both hepatocellular and cholestatic drug injury have a similar rate of mortality [35,36,58].
Female Gender.
There is also wide-spread belief that women are more likely than men to develop hepatotoxicity, especially with the use of herbal products or dietary supplements. The reasons offered for this include the lower body and liver weight of women, the greater use of and compliance with medication by women, and possible differences in drug-metabolizing enzyme activities. However, this view has been challenged by several reports [35,36,59]. In an analysis of Spanish Hepatotoxicity Registry patients, no difference in rates was found between men and women [35]. Similarly, in the ongoing Drug Induced Liver Injury Network (DILIN) prospective study, the frequency of drug-induced liver injury has been similar in men and women, although hepatocellular liver injury has occurred more frequently in women than men (65 versus 35%) [36].
Women may not be more susceptible to developing drug-induced liver injury but they do seem at increased risk for adverse outcomes. Data from the Acute Liver Failure study show a preponderance of women with paracetamol-related fulminant hepatitis as well as more severe idiosyncratic drug-induced liver disease [37,38]. Also, data from the Spanish Hepatotoxicity Registry indicated that more women than men required liver transplantation or died as a result of hepatotoxicity [60].
Age.
The incidence of drug-induced liver injury is believed to increase with advancing age at exposure. The proposed reasons are that older people are more likely to use medications, especially multiple drugs, and that they are likely to have altered drug pharmacokinetics, accounting for reduced drug metabolism, distribution and elimination [61,62]. Some evidence supports an increased risk of hepatotoxicity with increasing age from drugs such as isoniazid and amoxicillin–clavulanate [63,64]. A study from Japan of 142 patients hospitalized for hepatotoxicity reported that persons older than 75 years of age were more likely than those under 75 to have higher levels of alkaline phosphatase and a cholestatic profile at presentation [65]. In addition, data from the Spanish registry indicates that subjects over age 60 are more likely to present with cholestasis [64]. Thus, older persons appear more prone than younger people to develop drug-induced cholestatic liver injury.
The frequency of hepatotoxicity is lower in children than adults, in part because they less commonly take medications, and rarely multiple drugs. Moreover, children appear less susceptible than adults to developing drug-induced liver injury, with a few exceptions. Children with viral infections have a relatively high predilection for developing Reye’s syndrome when treated with aspirin [66]. They also appear more likely to develop a similar pattern of liver injury from treatment with valproate and erythromycin [67]. These effects may relate to age-related differences in P-450 gene expression, which is known to be markedly increased in children compared to adults.
Other Potential Predictors.
Underlying chronic viral hepatitis, B or C, is believed to increase the risk of developing hepatotoxicity. This may be due to altered pharmacokinetics, up-regulated intrahepatic cytokine expression or alterations in drug metabolizing pathways. There are reports of an increased risk of drug-induced liver injury in persons with chronic viral hepatitis who are treated with isoniazid and rifampicin [68,69] as well as with ibuprofen and methimazole [70]. Also, there are multiple reports of an increased risk of hepatotoxicity in persons with human immunodeficiency (HIV) infection coinfected with hepatitis B or C treated with highly active antiretroviral therapy [71,72]. To further support a role for HCV infection is the evidence that the risk of hepatotoxicity is reported to decline with successful eradication of HCV RNA [71]. Additional prospective research of viral replication and immunological status is necessary, however, to ensure that the liver injury is not a result of viral flare.
Obesity and non-alcoholic fatty liver disease (NAFLD) do not appear to increase the risk of developing drug-induced liver injury. Numerous studies have shown that statins do not increase the risk of developing hepatotoxicity in persons with obesity and NAFLD [73–75]. Obesity is, however, associated with poorer outcomes in persons who develop acute liver failure, although the pattern, severity and outcome of drug-induced liver injury are not influenced by an increased body mass index measurement [35,58].
The poorer outcome may be the consequence of inapparent underlying obesity-related chronic liver disease, such as cirrhosis.
Alcohol.
The role played by alcohol in enhancing hepatotoxicity requires additional study. There are reports indicating that the risk of isoniazid hepatotoxicity is increased in alcoholics [76]. Also, much attention has focused on determining whether alcohol augments paracetamol hepatotoxicity [77–79]. Indeed, it is theoretically plausible that chronic alcoholism increases the risk of paracetamol hepatotoxicity because alcohol induces hepatic CYP2E1, the enzyme responsible for converting paracetamol to its toxic metabolite, and because chronic alcoholism is associated with a reduction in protective glutathione and other essential nutrients [80]. There are even reports that chronic alcoholism enhances hepatotoxicity from receipt of low doses of paracetamol [79]. Challenging this view are data from the Acute Liver Failure study that showed a similar frequency of alcohol use among those with paracetamol hepatotoxicity regardless of whether the drug overdose was intentional or non-intentional [81]. Furthermore, the DILIN study found that an absence of alcohol consumption was associated with a poorer outcome [39].
Mechanisms of Injury, Drug Metabolism and Pharmacokinetics
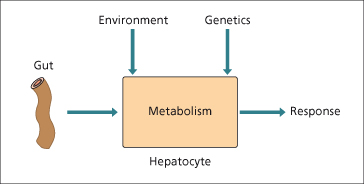
Hepatic clearance of drugs given orally depends upon the efficiency of drug-metabolizing enzymes, intrinsic clearance, liver blood flow and the extent of plasma protein binding (Fig. 24.2). Drugs vary in their pharmacological effects based on these different pharmacokinetic factors [61].
Fig. 24.2. The response of the liver to drugs depends on the interplay between absorption, environmental factors and genetics.
Hepatic Drug Metabolism
Phase 1.
The main drug-metabolizing system resides in the microsomal fraction of the hepatocyte, the smooth endoplasmic reticulum. Oxidation or hydroxylation of drugs, most commonly mediated through cytochrome (CYP) P450s, improves their solubility. Other drug-metabolizing reactions, such as conversion of alcohol to acetaldehyde by alcohol dehydrogenases, are present in the cytosolic fraction.
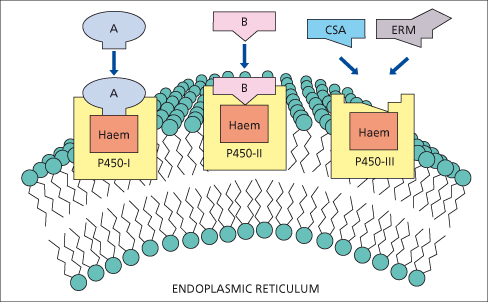
CYP enzymes are divided into gene families based upon 40% homology using Arabic numbers of 1 to 10; subfamilies are designated by a capital letter and specific isoforms are sequentially numbered. Currently, three CYP gene families (CYP1, 2 and 3) are identified as having a major role in drug metabolism (Fig. 24.3) [80]. Inducers of CYP metabolic pathways include barbiturates, alcohol, anaesthetics, hypoglycaemic and anticonvulsant agents, griseofulvin and rifampicin. Enzyme induction by these drugs may lead to slight liver enlargement.
Fig. 24.3. P450s involved in drug metabolism are members of three gene families: P450-1, P450-2 and P450-3. Individual P450s have distinct catalytic properties. Ciclosporin (CSA) and erythromycin (ERM) bind to and are metabolized by P450s within the P450-3 family [80].
Phase 2.
Conjugation of a drug or its metabolite with a small endogenous molecule serves to further enhance solubility and elimination from the body. The enzymes concerned are usually not confined to the liver but are present there in high concentration (i.e. glucuronidation, sulfotransferases, acetyltransferases).
Phase 3.
Active transport of drugs typically takes place at the biliary pole of the hepatocyte. Drug transport is energy dependent and can be saturated. Multiple factors determine whether the metabolized drug will be excreted in bile, urine or both. Highly polar parent drugs or metabolites as well as those with a molecular weight exceeding 200 Da are typically excreted in the bile. In contrast, the urinary route is more important with smaller molecules (Fig. 24.4). In humans, the multidrug resistant protein (MDR) is involved with transport of cationic drugs into bile. In addition, the multidrug resistance-associated proteins (MRPs) are involved in drug transport in the liver. The bile salt excretory protein (BSEP) and MDR3 are two other important canalicular transport proteins involved in drug excretion with a complex system involved in their regulation and function.
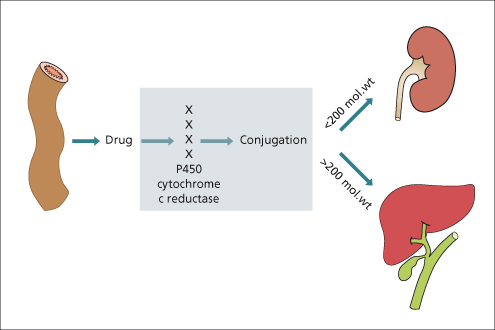
The Cytochrome P450 System
Each CYP protein is encoded by a unique gene which has variable expression among individuals, accounting in part for the fourfold or higher difference in drug metabolism among healthy persons. Each CYP isoform has a unique ‘substrate’ binding site capable of binding some, but not all, drugs. Inter-individual differences in the expression and translation of CYP proteins may determine idiosyncratic reactions to drugs. An example is the poor metabolism of debrisoquine (an antiarrhythmic drug) due to abnormal expression of CYP2D6 [82], which can be identified by PCR amplification of parts of the mutant genes of CYP2D6. CYP2E1 is involved in the production of electrophilic metabolites of paracetamol. CYP3A is concerned with the metabolism of ciclosporin and many other drugs including erythromycin, statins and ketaconazole. CYP2C polymorphism affects the metabolism of mephenytoin, diazepam and many other drugs.
Enzyme Induction and Drug Interactions.
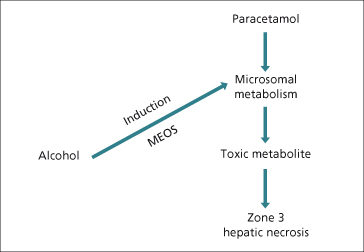
Induction of P450 enzymes may enhance the production of toxic metabolites. Ethanol induces CYP2E1 and may increase the toxicity of paracetamol via increased generation of N-acetylbenzoquinineimine (NAPQ1), a metabolite of paracetamol (Fig. 24.5). Similarly, paracetamol hepatotoxicity may increase in patients treated with isoniazid, which also induces CYP2E1 [83]. Ciclosporin, tacrolimus (FK506), erythromycin and ketoconazole compete for binding and metabolism by CYP3A4 and ciclosporin levels rise after they are given (Fig. 24.3).
Fig. 24.5. Alcohol, as an enzyme-inducing agent, increases the production of toxic metabolites of paracetamol (acetaminophen), thus potentiating hepatic necrosis. MEOS: microsomal enzyme oxidizing system.
Molecular Mechanisms in Drug-Induced Liver Injury
The clinical manifestations of drug-induced liver injury are a consequence of hepatocyte death mediated by either apoptosis or necrosis [84]. Apoptosis involves cellular shrinkage and fragmentation into discrete bodies which maintain intact plasma membranes (Fig. 24.6). These apoptotic bodies are rapidly cleared by phagocytosis, leaving little substrate to induce a host immune response. In contrast, necrosis involves a profound loss of mitochondrial function with ATP depletion, leading to cellular swelling and lysis which then promotes a local inflammatory response.
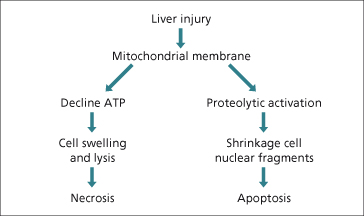
The disassembly process in apoptosis is executed by intracellular caspases (cysteine-dependent aspartate-specific proteases), existing in hepatocytes as zymogens. The caspase cascade is activated by initiators and terminated or perpetuated by other intracellular proteins or events. Increased mitochondrial permeability may arise from extrinsic activation of hepatocytes which occurs at the cellular surface (e.g. extrinsic pathway) or within the cell itself (intrinsic pathway). The principal hepatic death receptors are tumour necrosis factor-1 (TNF-1) and Fas [85]. Drugs and their metabolites may either induce TNF production by Kupffer cells or natural killer cells of the innate immune system or help sensitize hepatocytes to the effects of TNF.
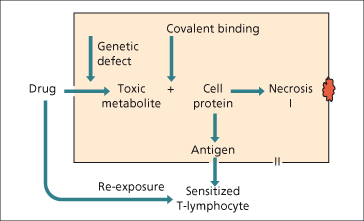
Drugs or their metabolites may also act as haptens and covalently bind hepatic proteins such as the CYP enzymes. Macrophages can then present these peptides on their cell surfaces bound to major histocompatability complex (MHC) class II. The adaptive immune system consisting of CD4 and CD8 T cells then contributes to apoptotic cell death mediated through Fas-ligand (Fig. 24.7). However, haptenization alone may be insufficient to trigger an immune response and a concomitant ‘danger’ signal such as mild background liver injury or a concomitant infectious or inflammatory condition may be required [86]. Important clinical correlates of intrahepatic and systemic danger signals include the increased risk of drug-induced liver injury and other adverse drug reactions in patients with AIDS [87].
Fig. 24.7. The mechanisms of hepatotoxicity, direct metabolite-related and immunological hypersensitivity.
Non-inflammatory or bland drug-induced cholestasis, as occurs with oral contraceptives or ciclosporin, may be due to inhibition of BSEP [88]. However, most clinically overt cholestatic reactions are associated with bile duct injury and a variable amount of inflammation. An hypothesis is that toxic drug metabolites undergo canalicular excretion and that exposed cholangiocytes are then injured as a result of an immune mediated reaction [89]. Interindividual differences in BSEP and MDR3 expression may play a role in drug-induced cholestasis [90].
Genetic Polymorphisms in Drug-Induced Liver Injury
Like other complex diseases, different genes and environmental factors may contribute to both susceptibility and outcome in drug-induced liver injury [91,92]. Genes involved may encode proteins with roles in several different pathways including drug metabolizing enzymes, drug transporters, apoptosis, acquired and innate immune responses and cellular repair and regeneration. Traditional studies have included candidate gene approaches with arbitrary selection of polymorphisms from genes of potential interest. For example, the slow acetylator genotype of N-acetyltransferase-2 (NAT-2) has been associated with an increased risk of isoniazid liver injury in some but not all studies [93–95]. Similarly, several studies have explored human leucocyte antigen (HLA) genotype polymorphisms and amoxicillin–clavulanate hepatotoxicity [96–98]. Two studies [96,97] involving 35 and 22 patients, respectively, identified a highly significant association of HLA DRB1*1501 with hepatotoxicity, a finding not confirmed in an independent cohort of 27 Spanish patients [98].
Commercial platforms containing probes for 500 000 and 1 000 000 single nucleotide polymorphisms (SNPs) typically present in 1 to 5% of the general population have also been proposed for genetic association studies of drug-induced liver injury [99]. With no specific a priori hypothesis, such extensive genotyping invariably identifies large numbers of associations by chance and therefore the threshold for significance in these studies is typically set at P = 10−7 to 10−8. Accordingly, it is now common to require duplication of a significant association in a second, independent cohort [100]. For example, the HLA-B*5701 haplotype was found to confer an 80-fold increased risk in susceptibility to flucloxacillin hepatotoxicity, a finding replicated in a second independent cohort of European patients [101]. However, since almost 5% of Caucasians have this haplotype, the absolute risk of developing drug-induced liver injury remains low in patients with this genotype (1 in 500). Nonetheless, knowing the HLA genotype could be helpful in establishing a diagnosis of drug-induced liver injury [102].
Diagnostic Approaches and Causality Assessment of Drug-Induced Liver Injury
Diagnosing hepatotoxicity with certainty is problematic for several reasons. First, hepatotoxicity can simulate virtually every type of liver disease so that, in the absence of biomarkers or other specific tests [103], drug-induced liver injury is at present a diagnosis of exclusion [84]. It is therefore mandatory when assessing potential hepatotoxicity to exclude all causes of liver injury that it can mimic. Second, having access to all sequential clinical and biochemical data related to the injury, which is not always available, is key to defining the characteristics and pattern of the liver injury which aids in its diagnosis. Third, the medical condition for which the drug is given may itself cause liver dysfunction and thus confound the adjudication process. Fourth, because multiple drugs are commonly used, synergistic interactions may result as well as uncertainty about which drug is actually responsible for the injury. Finally, locating historical information that supports the potential for hepatotoxicity of a given drug can be challenging, especially in view of the innumerable drugs and herbals currently on the market.
Two primary methods are employed to assess causality in drug-induced liver injury—the use of general clinical acumen and utilization of numerical scoring systems [104,105]. The former involves applying careful judgment when clinical or biochemical evidence of liver injury appears linked to the use of a drug or herbal product. However, lacking a specific diagnostic test for hepatotoxicity, this approach to causality assessment is highly subjective, its accuracy depending upon the expertise of the interviewer and the intensity with which alternate causes are eliminated. Nevertheless, it is generally accepted as the ‘gold standard’ diagnostic approach; when undertaken by groups of experienced clinicians, the assessment is considered to be by ‘expert opinion’.
A more objective means of assessing causality was proposed by international experts at a meeting of the Council for International Organizations of Medical Scientists (CIOMS) in 1989 [106,107]. The result was the development of a structured numerical scoring system to grade the likelihood of drug-induced liver injury. The meeting was supported by Roussel-Uclaf Pharmaceuticals, and hence the instrument was called RUCAM (Roussel-Uclaf Causality Assessment Method). This instrument is now regularly employed by field experts in hepatotoxicity and by pharmaceutical companies to evaluate suspected cases of drug-induced liver injury, but it is difficult to use and therefore has not achieved wide clinical acceptance, even among practicing hepatologists.
Other grading instruments proposed have included the M and V system (clinical diagnostic scale) and the Naranjo scale [108–110]. However, the former has been found to be less effective than RUCAM [111], as has the latter [112] which was developed originally for assessing any adverse drug reactions.
Clinical and Biochemical Presentations of Drug-Induced Liver Disease
Clinical Presentation.
Hepatocellular injury presents similarly to acute viral hepatitis, symptoms ranging from none at all to those typical of acute fulminant hepatitis. Generally, symptoms are non-specific, consisting of fatigue, myalgia, nausea and/or vomiting and, occasionally, mild right upper quadrant discomfort or pain. Depending upon the drug, symptoms may include mild fever, arthralgias and pruritus. Jaundice, together with marked pruritus, is a hallmark of cholestatic liver injury.
Biochemical Presentation.
Generally, the first indicator of drug-induced liver injury is detection of raised serum ALT and aspartate aminotransferase (AST) levels in hepatocellular injury and the alkaline phosphatase in cholestatic injury. The enzyme pattern with hepatocellular drug-induced liver injury resembles acute viral hepatitis, including an ALT increase that exceeds the AST increase. An uncertain issue is what the level of ALT elevation should be that triggers withdrawal of the potentially offending drug. This decision is usually based on the severity of the liver damage, the gravity of the disease being treated, and whether or not there is an alternative replacement drug. Some discontinue the drug if the ALT level exceeds three times the upper limit of normal (ULN) on two or more occasions, others when it exceeds five times ULN, and still others when it exceeds eight times ULN [113]. However, the level of ALT increase does not necessarily define current liver disease severity nor is it necessarily predictive of outcome [113]. A more robust indicator of liver disease severity is the addition of hyperbilirubinaemia (total bilirubin ≥2.5 mg/dL) or of overt jaundice, a combination that has been associated with a 10% or greater risk of liver-related mortality [35,58]. The US Food and Drug Administration (FDA) refer to this as ‘Hy’s rule’, named for Dr Hyman J. Zimmerman, a pioneer in the study of hepatotoxicity [15,114]. Thus, if an affected person has debilitating clinical symptoms and/or increased levels of the total serum bilirubin, the drug should be discontinued even if the ALT elevation is only threefold elevated. Alkaline phosphatase is generally only slightly elevated in persons with hepatocellular injury, but is moderately to markedly increased with cholestatic injury. Bilirubin elevation is more common in cholestatic than in hepatocellular injury, but without necessarily having the same implications. This presentation, referred to as intrahepatic cholestasis, must be distinguished from jaundice of extrahepatic obstruction using imaging (e.g. ultrasonography) and/or other procedures (e.g. magnetic resonance cholangiopancreatography). Some drugs, such as oral contraceptives, may induce pure cholestasis, characterized by an increase only in the serum bilirubin level [88,115]. Serum enzymes less commonly assessed are γ-glutamyl transpeptidase and lactic dehydrogenase, although they can be moderately helpful in diagnostic evaluation.
Drug-induced liver disease may also present as mixed hepatocellular/ cholestatic liver disease, with significant increases in levels of both the aminotransferases and the alkaline phosphatase. Characterizing the presenting pattern of injury is important because drugs tend to be consistent in the type of injury they cause. However this is not always the case, because there may occasionally be deviation from the usual pattern or the pattern may change during the course of the injury, transitioning from hepatocellular to a mixed and even a cholestatic form over time [35].
For some drugs, the target is intracellular organelles such as mitochondria. Biochemical dysfunction in this situation ranges from mild to moderate elevations in bilirubin and aminotransferase levels to severe liver injury associated with an increase in the serum ammonia level and lactic acidosis.
Other Laboratory Tests.
Other important tests are the prothrombin time (or INR), the complete blood count and differential, and tests for autoantibodies. The prothrombin time helps define disease severity, identifying eosinophilia helps to establish the injury as immunoallergic, while identifying increased levels of ANA, antismooth muscle antibodies (SMA) and antimicrosomal antibodies helps categorize it as of autoimmune origin. Finding antimitochondrial antibodies (AMA) helps distinguish drug-induced cholestatic liver disease from primary biliary cirrhosis, although transient AMA positivity may rarely be seen with hepatotoxicity, particularly if fulminant hepatitis develops.
Histological Presentations.
A biopsy is commonly not performed because it is problematic to routinely require an invasive procedure if there are no specific interventions to offer, and the biopsy may not be specifically diagnostic for drug-induced liver injury. While there are histological features that strongly suggest drug injury, such as zonal necrosis, eosinophilia and microvesicular steatosis, the biopsy is often most useful for identifying alternative diagnoses, including unanticipated cirrhosis, given the near epidemic of non-alcoholic fatty liver disease. Nevertheless, an astute and experienced pathologist who couples histopathology with clinical findings can provide extremely helpful diagnostic and at times, prognostic information [42,43,116].
Assessment of Suspected Drug-Induced Liver Disease (Table 24.5)
The first step in evaluating a suspected case of hepatotoxicity is to exclude other more common causes of liver disease. With hepatocellular injury, conditions requiring exclusion are acute viral hepatitis, pre-existing autoimmune hepatitis, alcoholic liver disease, non-alcoholic fatty liver disease, acute congestive heart failure or other causes for hypotension [104,105]. Virus testing must include assays for hepatitis A (IgM anti-HAV), hepatitis B (HBsAg or IgM anti-HBc), and hepatitis C (anti-HCV and HCV RNA), and, preferably, hepatitis E, Epstein–Barr virus and cytomegalovirus infections. Measuring serum globulin levels and testing for autoantibodies helps exclude autoimmune hepatitis, while ultrasonography and cross-sectional imaging is used to screen for fatty liver and pancreaticobiliary disease such as gallstones or malignancy that may present with obstructive jaundice. Finally, complete evaluation requires excluding haemochromatosis, Wilson’s disease and α-1-antitrypsin deficiency.
Table 24.5. Evaluation of potential drug-induced liver injury
| History | All drugs and herbals used in past 12 months |
| Start and stop date(s) for all medications | |
| Past history of hepatotoxicity and use of implicated drug | |
| Symptoms at onset (fever, rash, fatigue, abdominal pain) | |
| Disease(s) for which medication taken | |
| Other disease(s), particularly cardiovascular | |
| Episode of acute hypotension | |
| Alcohol intake | |
| Physical examination | Jaundice, rash, clinical evidence of pruritis |
| Liver and spleen size | |
| Stigmata of chronic liver disease | |
| Routine chemistries | Complete blood count and differential, platelets, total protein, albumin/ globulin |
| Prothrombin time/ INR, creatinine | |
| Liver chemistries | alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, total/ direct bilirubin, γ-GTP |
| Serologies | IgM anti-HAV, HBsAg, anti-HBc IgM, anti-HCV, HCV RNA, anti-HEV, anti-EBV, anti-CMV |
| Autoantibodies | antinuclear antibody, smooth muscle antibody, antimitochondrial antibody |
| Special tests | Serum iron, ferritin, ceruloplasmin, α-1-antitrypsin |
| Imaging | Ultrasound, CT, MRI (ERCP) |
ERCP, endoscopic retrograde cholangiopancreatography; γ-GTP, γ-glutamyl transpeptidase.
Important historical information includes alcohol use, cardiovascular disease and whether there had been an episode of acute hypotension. These factors may lead to hepatocellular injury, although their injury patterns are reasonably distinctive and separable from drug-induced liver injury.
With no alternative explanation for acute liver injury but receipt of a drug, the next step is to consider which drug might be responsible and to review the circumstances surrounding and features of the liver injury. A careful history must be taken of all medications (e.g. prescription, herbal, weight loss) received in the preceding 12 months, including their precise start and stop dates. Also important is whether there had been a previous episode of hepatotoxicity and, if so, what drug had been implicated. Defining features of the liver injury is helpful in implicating specific medications since most drugs that cause liver injury have a characteristic ‘signature’. Requirements include: establishing the time interval between start of the drug and onset of liver disease (latency); defining the clinical pattern of the liver injury (hepatocellular, cholestatic, mixed); and recording the time to recovery after drug discontinuation (de-challenge). Also important is to determine whether the drug responsible for the current injury had been received in the past, thus representing a re-challenge. The clinical pattern is defined using the R value equation [106,107]:
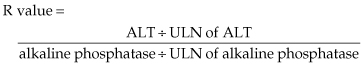
An R value ≥ 5 establishes hepatocellular; ≤2, cholestatic; and >2 but <5, a mixed pattern of injury.
Assessing Causality for Drug-Induced Liver Disease
Assessment Using Clinical Judgment.
Once a specific drug is identified as potentially responsible for the liver injury, the final steps are to grade it for the likelihood of its implication as well as the severity of the liver injury. Various grading systems have been devised for this purpose. The US DILIN study group has developed enhanced grading systems for assessing both the likelihood and severity of the liver injury, as shown in Tables 24.6 and 24.7 [117].
Table 24.6. The Drug Induced Liver Injury Network (DILIN) grading system for causality assessment
| Causality | Likelihood score (%) | Description |
| 1 Definite | >95 | Liver injury typical for drug or herbal; causality ‘beyond a reasonable doubt’ |
| 2 Highly likely | 75 to 95 | Causality ‘clear and convincing’ but not definite |
| 3 Probable | 50 to 74 | Causality supported by ‘the preponderance of the evidence |
| 4 Possible | 25 to 49 | Causality not supported by ‘the preponderance of the evidence’ |
| 5 Unlikely | <25 | Causality ‘highly unlikely’ |
| 6 Insufficient evidence | Not applicable |
Adapted from [117].
Table 24.7. The Drug Induced Liver Injury Network (DILIN) grading system for liver disease severity
| Score | Grade | Definition |
| 1 | Mild | ALT and/or alkaline phosphatase levels increased but total serum bilirubin <2.5 mg/dL and INR <1.5 |
| 2 | Moderate | ALT and/or alkaline phosphatase levels increased and total serum bilirubin ≥2.5 mg/dL or INR ≥1.5 |
| 3 | Moderate–Severe | ALT, alkaline phosphatase, bilirubin and/or INR increased and patient is hospitalized or ongoing hospitalization is prolonged because of drug-induced liver injury |
| 4 | Severe | ALT, alkaline phosphatase and total serum bilirubin is increased together with at least one of the following: (i) hepatic failure (INR ≥1.5, ascites or encephalopathy); or (ii) other organ failure due to liver injury |
| 5 | Fatal | Death or liver transplantation because of drug-induced liver injury |
ALT, alanine aminotransferase.
Adapted from [117].
Follow-Up of Acute Drug-Induced Liver Injury.
It is important to follow the affected person to resolution of the clinical symptoms and biochemical abnormalities. This provides important de-challenge information as well as information on whether the injury advanced to chronic liver disease [116]. Liver biopsy examination at this time helps categorize the status of the liver disease.
Assessment Using RUCAM.
The RUCAM, designed as an objective measure, confers points for clinical, biochemical, serological and radiological characteristics of liver injury [106,107]. Seven domains are included in the system:
- time to onset of the liver disease after starting the drug
- course of the liver disease
- risk factors for developing liver injury
- potential for hepatotoxicity of concomitant drugs
- exclusion of non-drug causes of liver injury
- previous information regarding hepatotoxicity of the implicated drug
- response to re-administration (i.e. re-challenge).
Each domain is awarded a positive or negative numerical score, the total ranging from −9 to +14. The scoring components differ somewhat according to the pattern of liver injury (hepatocellular, cholestatic/ mixed).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


