Prehepatic.
This may be due to an increased load of bilirubin arriving at the liver, as in haemolysis, or diminished hepatic capacity for conjugation of bilirubin, as in Gilbert’s and Crigler–Najjar syndrome. The circulating serum bilirubin is largely unconjugated and the serum transaminase and alkaline phosphatase are normal. Bilirubin cannot be detected in urine.
Hepatic.
Hepatocyte damage results in reduced efficiency of bilirubin excretion into bile. Conjugated bilirubin refluxes into the circulation and is found in urine. Serum biochemistry shows an increase in hepatic enzymes according to the underlying cause.
Cholestatic.
This is due to failure of adequate amounts of bile to reach the duodenum, either through a specific failure of canalicular secretion or physical obstruction to bile flow at any level. The serum shows increases in conjugated bilirubin, biliary alkaline phosphatase, γ-glutamyl transpeptidase (γ-GT), total cholesterol and conjugated bile acids. Lack of bile acids to form micelles may cause steatorrhoea and malabsorption of calcium and the fat-soluble vitamins A, D, E and K.
Physiology and Pathophysiology
Bilirubin Metabolism and Transport
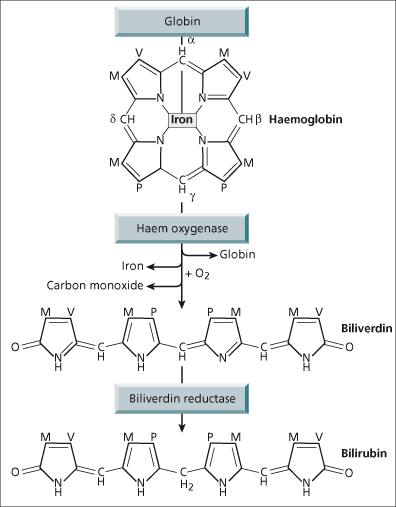
Bilirubin is an end-product of haem catabolism, the majority (80–85%) of which is derived from haemoglobin and only a small fraction from other haem-containing proteins such as cytochrome P450 (Fig. 11.2), myoglobin and immature bone marrow cells. Approximately 300 mg bilirubin is formed daily.
The enzyme that converts haem to bilirubin is microsomal haem oxygenase (Fig. 11.3). Cleavage of the porphyrin ring occurs selectively at the α-methane bridge. The α-bridge carbon atom is converted to carbon monoxide and the original bridge function is replaced by two oxygen atoms which are derived from molecular oxygen. The resulting linear tetrapyrrole has the structure of IX α-biliverdin, which is converted to IX α-bilirubin by biliverdin reductase, a cytosolic enzyme. Such a linear tetrapyrrole should be water soluble, but realignment of the pyrrole ring allows internal hydrogen bonding to mask the propionic acid side chains and thus make bilirubin poorly soluble in aqueous solvents. The bonding is broken by alcohol in the diazo (van den Bergh) reaction converting unconjugated, indirect bilirubin to direct reacting bilirubin. In vivo hepatic esterification of bilirubin’s propionic groups by glucuronic acid makes it water soluble. Unconjugated bilirubin is non-polar (lipid soluble) and transported in the plasma tightly bound to albumin. Only a very small amount is dialysable, but this can be increased by substances such as fatty acids, organic anions and drugs, which compete with bilirubin for albumin-binding sites. Avoidance of such competitive binders is vitally important in the neonate when diffusion of unbound bilirubin into the brain can cause kernicterus [1].
Bilirubin is efficiently removed from albumin and internalized by the liver where it is bound initially to cytosolic carrier proteins such as glutathione-S-transferase (ligandin), then glucuronidated and excreted into bile.
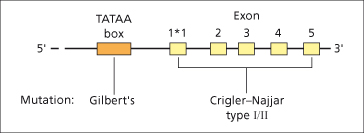
Microsomal bilirubin uridine diphosphate glucuronosyl transferase (UGT) is the enzyme that converts unconjugated bilirubin to conjugated bilirubin mono- and diglucuronide. The gene expressing bilirubin UGT is on chromosome 2. The structure of the gene is complex (Fig. 11.4) [2]. Exons 2–5 at the 3′ end are constant components of all isoforms of UGT but, to complete their expression, one of several first exons can be employed. Each exon 1 sequence has different substrate specificity and enzyme characteristics. Exon 1*1 encodes the variable region for bilirubin UGT1*1, responsible for virtually all conjugation of bilirubin to both its monoglucuronide and diglucuronide. The major conjugate in human bile is the diglucuronide, though sulphate, xylose and glucose conjugation also occur to a small extent and may be increased in cholestasis. Biliary excretion of the glucuronide is mediated by the ATP-dependent multidrug resistance protein-2 (MRP-2) [3,4] and is the rate-limiting factor in the transport of bilirubin from plasma to bile.
Fig. 11.4. Structure of the gene for bilirubin UGT1*1 with five exons and the promoter region (TATAA box). There are several other possible first exons (not shown) that can be spliced to exons 2–5, and have other substrate specificities.
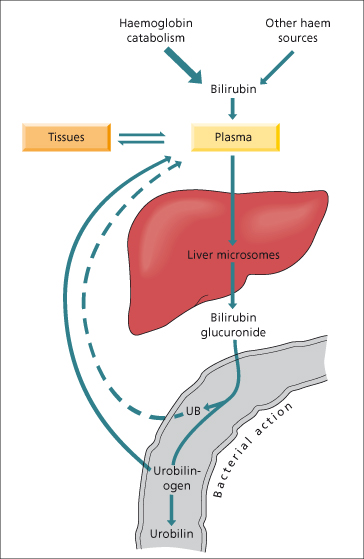
Bilirubin diglucuronide is not reabsorbed from the small intestine but in the colon may be hydrolysed by bacterial β-glucuronidases, producing urobilinogens and urobilin which are excreted in the stool or urine. In cholangitis, bacterial hydrolysis of bilirubin glucuronide in the biliary tree produces unconjugated bilirubin which may result in production of pigment gallstones.
Secretion of Bile
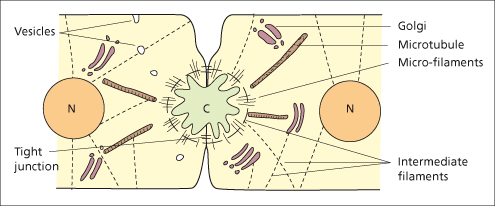
Bile is produced by hepatocytes and modified by cholangiocytes lining the bile ducts. Bile formation is dependent on energy-dependent transport processes in the basolateral and canalicular membranes of the hepatocyte and cholangiocytes and largely independent of perfusing blood pressure. Conjugated bilirubin, bile salts, cholesterol, phospholipids, proteins, electrolytes and water are secreted by the liver cell into the canaliculus. The bile secretory apparatus comprises the canalicular membrane with its carrier proteins, the intracellular organelles and the cytoskeleton of the hepatocyte. Tight junctions between hepatocytes seal the biliary space from the blood compartment (Fig. 11.5).
Fig. 11.5. The biliary secretory apparatus. Diagram of the ultrastructure of the bile canaliculus (C), cytoskeleton, and organelles (N, nucleus).
The canalicular membrane contains carrier proteins which transport bile acids, bilirubin, cations and anions (Fig. 11.6). Canalicular secretion is modified by water and electrolytes passing between hepatocytes across the tight junction (paracellular flow) in response to osmotic gradients between canalicular bile and the intercellular fluid which is in continuity with the space of Disse [5]. Canaliculi empty their bile into ductules, sometimes called cholangioles or canals of Hering, which connect with interlobular bile ducts, the first bile channels to be accompanied by a branch of the hepatic artery and portal vein to form the triad which is characteristically found in portal tracts. Cholangiocytes, whose properties differ between small and large ducts, contribute a further 150 mL/day [6,7].
The passage of conjugated bile salts into the biliary canaliculus is the most important factor promoting bile formation. Water follows the osmotically active bile salts and there is a tight relationship between bile flow and bile salt secretion. Total bile flow in man is about 600 mL/day. The hepatocyte provides two components: bile salt dependent (approximately 225 mL/day) and bile salt independent (approximately 225 mL/day). When bile flow is plotted against bile salt secretion, bile salt independent flow is the value obtained when bile salt excretion is extrapolated to zero. In this case other osmotically active solutes, such as glutathione and bicarbonate, generate water flow.
Cellular Mechanisms
Sinusoidal Uptake
Bile formation requires the uptake of bile acids and other organic and inorganic ions from plasma across the basolateral (sinusoidal) membrane, transport through the hepatocyte and excretion across the canalicular membrane (Fig. 11.6). Na+/K+–ATPase, which is found only on the basolateral membrane, exchanges three intracellular sodium ions for two extracellular potassium ions, thus maintaining the sodium (high outside : low inside) and potassium (low outside : high inside) gradients. In addition, because of the imbalance of electrical exchange, the cell interior is negatively charged (−35 mV) compared with the exterior, favouring uptake of positively charged ions and excretion of those with a negative charge.
Fig. 11.6. Major transport systems in bile formation. Note the Na+/K+ ATPase or sodium pump (centre top), the sinusoidal Na+ taurocholate co-transporting protein (NTCP), the sinusoidal multispecific organic anion transporter (OATP) and the organic cation transport 1 (OCT1). The canalicular membrane transporters are: BSEP, the bile salt export pump; MRP2, the multispecific organic anion transporter; MDR1, the ATP-dependent transporter of organic cations; and MDR3, an ATP-dependent phospholipid transporter (flippase). Other transport systems include a sinusoidal Na+–H+ exchanger, and canalicular bicarbonate transporter.
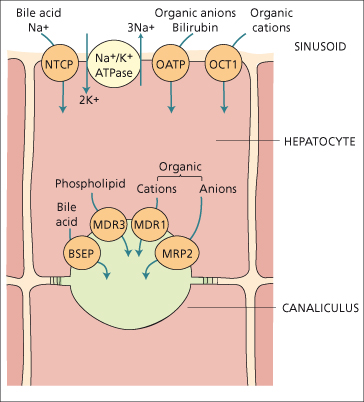
The sodium gradient drives the Na+-dependent taurocholate cotransporter protein which internalizes plasma bile acids conjugated with taurine or glycine. The organic anion transporter protein is sodium independent and carries several molecules, including bile acids, bromsulphthalein and other organic anions. There is also an organic cation transporter. Less well-defined carriers are thought to transport bilirubin into the hepatocyte [8].
Other ion transporters on the basolateral surface are the Na+–H+ exchanger involved in control of intracellular pH. A Na+–HCO3 cotransporter also serves this function. The basolateral membrane also contains uptake processes for sulphate, non-esterified fatty acids and organic cations.
Canalicular Secretion (Figs 11.6, 11.7, 11.8)
The canalicular membrane, which accounts for only 1% of the hepatocyte’s surface area, contains transporters [10] which secrete molecules into bile against steep concentration gradients as well as enzymes such as alkaline phosphatase and γ-GT. The transporters mainly belong to the family of ATP-binding cassette (ABC-) proteins, of which several hundred have been identified across many organisms. Canalicular MRP-2 (a member of the multidrug resistance protein family) excretes glucuronide and glutathione-S-conjugates, for example bilirubin diglucuronide [8]. The canalicular bile salt export pump [11] carries bile acids and is in part driven by the negative intracellular electric potential.
Fig. 11.7. Specialized domains of the hepatocyte membrane are illustrated by freeze fracture (a,c,d). On transmission electron microscopy (b) the basolateral intercellular space is marked with an electron dense marker (white arrow). In (a) the bile canaliculus (BC) can be seen running along the surface of a trabeculum of hepatocytes whose basolateral surfaces are in contact with blood within the sinusoids (S) on each side. (c) The sinusoidal endothelial cell is fenestrated, allowing the microvilli of hepatocytes (arrows) to come into direct contact with blood. (d) The canalicular and basolateral membrane domains are separated by tight junctions (zonula occludens, black arrows). Canalicular microvilli are indicated by white arrows. The inner and outer surfaces of the basolateral membrane of adjacent hepatocytes are marked.

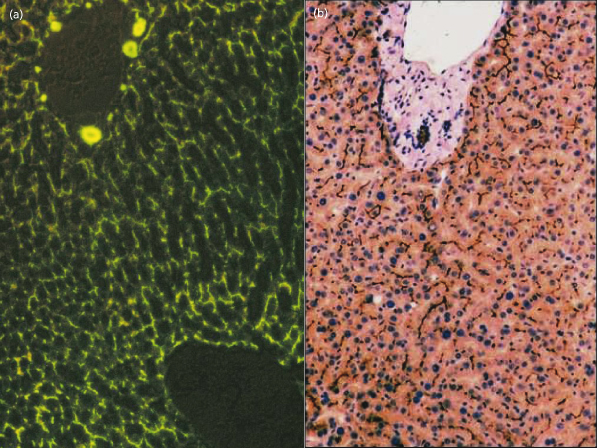
Fig. 11.8. (a) Bile flow is made visible within the canaliculi by the presence of the fluoroscein-conjugated bile acid, cholyl lysyl fluoroscein [9]. (b) Immunostaining (dark brown) by antibodies to the bile acid transporter (BSEP) show its expression to be confined to the canalicular membrane.
Two members of the P-glycoprotein family are important in canalicular transport; both are ATP dependent. MDR1 (multiple drug resistance 1) is a transporter of hydrophobic organic cations, and derives its name from being responsible for transporting cytotoxic drugs out of cancer cells, rendering them resistant to these drugs. The endogenous substrate is not known. MDR3 is a phospholipid flippase, which selectively enriches the canalicular (outer) surface of the membrane bilayer with phosphatidyl choline (lecithin) from where it is leeched by bile acids to become the major phospholipid in bile [12]. Hepatocytic active transport of the various biliary solutes into the canaliculus creates the osmotic and electrochemical gradients which result in diffusion of water and inorganic ions (in particular sodium) into the canaliculus. The rate of bile secretion is influenced by many hormones and second messengers including cyclic AMP and protein kinase C. Passage of bile along the canaliculus involves the action of contractile microfilaments.
Ductular Modification of Bile
Cholangiocytes lining the larger bile ducts participate in hormone-regulated ductal secretion of a bicarbonate-rich solution, so-called ductular bile flow. They express secretin receptors, the cystic fibrosis transmembrane conductance regulator (CFTR), the chloride–bicarbonate exchanger and somatostatin receptors. Interaction of secretin with its receptor increases intracellular cyclic AMP synthesis and activates protein kinase A (PKA) which activates the chloride channel (CFTR). Function of the chloride– bicarbonate exchanger depends upon the transport of chloride ions by CFTR into the bile duct. Chloride within the bile duct lumen is reabsorbed into cholangiocytes in exchange for bicarbonate [13]. Interaction of somatostatin with its receptor (SSTR2) on the basolateral surface of large cholangiocytes depresses cyclic AMP synthesis, a reversal of the above [7]. Bombesin and vasoactive intestinal peptide (VIP) increase bile flow through stimulation of the chloride–bicarbonate exchanger. Gastrin, insulin and endothelin inhibit secretin-induced bicarbonate-rich choleresis. Acetyl choline increases basal- and secretin-stimulated bicarbonate secretion. Secretin triggers the insertion of aquaporin 1 into the apical membrane of the cholangiocyte and this facilitates transport of water into bile. Aquaporin 4, in the basolateral membrane, subserves entry of water into the cell [14].
Ursodeoxycholic acid and other bile acids with a pKa which is sufficiently high that it enables them to exist within bile in their non-ionized (protonated) state, may be reabsorbed across the biliary epithelium by non-ionic diffusion. Such bile acid ‘short circuits’ from bile duct to liver (‘cholehepatic shunting’) explain the choleretic effect and high biliary bicarbonate secretion associated with ursodeoxycholic acid and nor-bile acids [15].
Bile Acid Metabolism and Transport
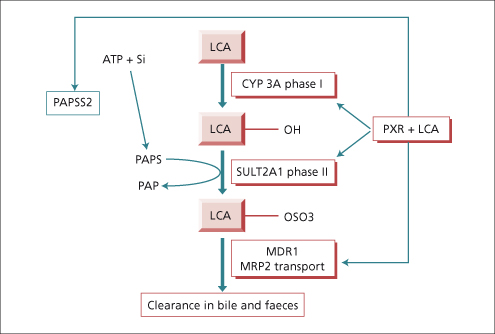
The human liver synthesizes the primary bile acids—cholic acid (3α,7α,12α trihydroxy cholanoic acid), chenodeoxycholic acid (3α,7α dihydroxycholanoic acid) and a little (1%) ursodeoxycholic acid, the 7β isomer of chenodeoxycholic acid (see Chapter 2). Their dehydroxylation by bacteria in the intestine produces deoxycholic acid (3α,12α dihydroxy) and lithocholic acid (3α monohydroxy), known as secondary bile acids. The liver conjugates both primary and secondary bile acids with either glycine or taurine. The bile acids circulate from liver to intestine and back about 6–10 times per day, about 5% being replaced by de novo hepatic synthesis to compensate for their loss in faeces. Provided bile acid loss in faeces does not exceed 20%, a constant bile acid pool size is sustained. This is through feedback regulation of de novo bile acid synthesis by the farnesoid X receptor as well as by cytokines and by a peptide (FGF-19) liberated by bile acids from the ileal enterocyte [15,16]. Protection against the potentially severe toxicity of lithocholic acid is coordinated by the pregnane X receptor which has the potential to switch on all the genes required for its safe metabolism and elimination, including hydroxylation by cytochrome P450 3A, sulphation by sulphotransferase (SULT2A1) and transporter promoted excretion in faeces [17,18] (Fig. 11.9).
Fig. 11.9. The pregnane X receptor (PXR) is the hepatocyte’s ligand for the potentially toxic secondary bile acid, lithocholic acid (LCA) as well as a vast array of other toxic substances. The nuclear receptor coordinates enhanced synthesis of the substrate donor (3′-phosphoadenosine 5′-phosphosulfate synthetase, PAPS) for sulphation, expression of the enzymes required in hydroxylation and sulphation of LCA, (phase I and II biotransformation) as well as expression of the active transport pumps which eliminate the metabolites in bile. CYP 3A, cytochrome P450 3A; SULT 2A1, sulphotransferase 2A1; MDR, multidrug resistance protein.
Syndrome of Cholestasis
Definition
Cholestasis is impaired formation or flow of bile. This can occur at any level between the basolateral (sinusoidal) membrane of the hepatocyte and the ampulla of Vater.
Morphologically, cholestasis is recognized from accumulation of biliary constituents in liver cells and biliary passages.
Clinically, cholestasis results in retention in blood of substances normally excreted in bile typically with elevated levels of serum alkaline phosphatase (biliary isoenzyme), γ-GT and cholesterol. Itching (pruritus) is the most typical symptom but not always present. Bilirubin darkens the urine. Lack of sufficient bile acids for micellar solubilization of the products of intestinal fat digestion causes steatorrhoea and may result in deficiencies of the fat-soluble vitamins A, D, E and K which can not be absorbed in the absence of bile acids.
Classification
Cholestasis may be classified as intra- or extrahepatic, and acute or chronic.
Clinical Features
Jaundice is not a constant finding in cholestasis. Patients have been treated for a decade or more on the basis of a mistaken diagnosis of dermatological disease when pruritus was not accompanied by jaundice. The sclerae are yellowed in jaundice but not by other causes of yellowing of the skin. The basal ganglia may be stained yellow in the newborn (kernicterus) due to the high concentration of circulating unconjugated bilirubin having an affinity for neural tissue. Circulating protein-bound bilirubin does not easily enter protein-low tissue fluids; thus exudates tend to be more icteric than transudates. The extremely rare symptom of xanthopsia (seeing yellow) is typically due to deficiency of vitamin A caused by the cholestasis and resolves within a few hours of parenteral administration of vitamin A.
Typically, the cholestatic patient feels well. The liver is usually enlarged with a firm smooth non-tender edge. Splenomegaly is unusual except in biliary cirrhosis where portal hypertension has developed. Stools become pale, their colour giving a good indication of whether cholestasis is total, intermittent or decreasing. Prominent features of cholestasis, both acute and chronic, are itching and malabsorption of fat and fat-soluble nutrients. Hence, chronic cholestasis is associated with bone disease (hepatic osteodystrophy), cutaneous deposition of cholesterol (xanthoma, xanthelasma) and darkening of the skin by melanin. In steatorrhoea stools are loose, pale, bulky and offensive, mandating a reduction of dietary fat intake to prevent associated weight loss; caloric substitution can be achieved with medium-chain triglycerides as they are absorbed directly into the portal vein, by-passing the requirement for lipolysis, micelle formation and lymphatic chylomicron transportation which are essential components of long-chain triglyceride assimilation (Table 11.1).
Table 11.1. Dietary management of the cholestatic patient
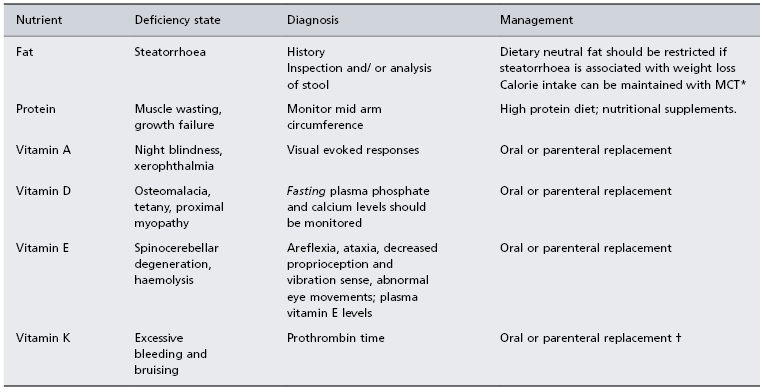
* MCT: medium-chain triglycerides are absorbed into the portal vein, thus by-passing the need for digestion, micelle formation and lymphatic transport which are essential for long-chain triglyceride assimilation. MCT can be given as Liquigen (Scientific Hospital Supplies Ltd, UK) or as MCT (coconut) oil for cooking or in salads.
† Parenteral vitamin K (10 mg) should correct prothrombin time within a day if prolongation due to deficiency alone.
Itching (Fig. 11.10).
Itching is typically mild before breakfast and at its worst in the evenings, hindering sleep. Night-time fasting permits catch-up concentration of biliary elements in the gallbladder, but during the day they re-accumulate in the systemic circulation due to impaired hepatic clearance of each meal-stimulated enterohepatic cycle of bile. The anion-binding resin cholestyramine is therefore most efficacious if present in the duodenum during vigorous gallbladder emptying stimulated by a good breakfast. The association of pruritus with cholestasis suggests that it is caused by a substance normally excreted in bile, and the efficacy of cholestyramine in providing relief from pruritus strongly suggests that the pruritogenic factor has an enterohepatic circulation. Nevertheless, the severity of pruritus does not correlate with the concentration of any naturally occurring bile acid in serum or skin [19]. Disappearance of itching when end-stage liver failure ensues suggests that the agent responsible is manufactured by the liver. Despite these clues as to its identity the exact cause of pruritus remains unknown. Attention has turned towards agents that may produce itching by a central neurotransmitter mechanism [20]. Opiate agonists induce opioid receptor mediated scratching activity of central origin. Cholestatic animals have evidence of increased opioidergic tone, reversible by naloxone. Opiate antagonists reduce scratching in cholestatic patients [21] and may produce opioid withdrawal-like reactions.
Fig. 11.10. A patient with cholestatic jaundice who demonstrates the classical features of (a) cutaneous cholesterol deposits and (b) scratch marks in response to pruritus.
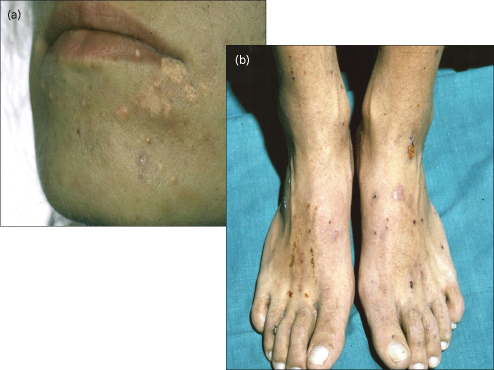
Fatigue.
This is troublesome in 70–80% of patients with chronic cholestatic liver disease. Experimental data show behavioural changes in cholestasis and suggest a central mechanism involving serotoninergic neurotransmission and/or neuroendocrine defects in the corticotrophin-releasing hormone axis [22].
Fat-Soluble Vitamins [23] (Table 11.1).
Before any invasive techniques are embarked upon for investigation and treatment of cholestasis, it may be necessary to administer vitamin K parenterally to correct the prolonged prothrombin time. In prolonged cholestasis, plasma vitamin A levels fall due to poor absorption, and hepatic reserves may become exhausted, resulting in failure of dark adaption (night blindness). Vitamin D deficiency may lead to osteomalacia. Spinocerebellar degeneration has been reported in adolescents with cholestasis and cystic fibrosis-induced vitamin E deficiency [24]. The picture is of cerebellar ataxia, posterior column dysfunction, peripheral neuropathy and retinal degeneration. If the serum bilirubin level exceeds 100 µmol/L (6 mg/dL) almost all adult patients with cholestasis will have subnormal vitamin E levels.
Xanthomas.
Flat or slightly raised yellow skin deposits are usually noted around the eyes (xanthelasma) (Fig. 11.10) but may also appear in palmar creases, below the breast and on the neck, chest or back. Tuberous (nodular) lesions are found on extensor surfaces, on pressure points and in scars. Cholesterol deposits may regress and disappear during treatment with statins, following resolution of cholestasis or with advancing hepatocellular failure.
Hepatic Osteodystrophy [23] (Table 11.2)
Bone disease is a complication of chronic liver disease, particularly chronic cholestasis. Untreated, it can lead to bone pain and fractures. Studies show that osteoporosis is responsible for the bone changes in the majority of patients with primary biliary cirrhosis and primary sclerosing cholangitis, although the potential for osteomalacia also exists. In osteoporosis there is loss of bone, both matrix and mineral. In osteomalacia there is defective mineralization of osteoid. Risk factors for osteoporosis include low body mass index, steroid treatment, increasing age and female sex [25]. One-third of patients with primary biliary cirrhosis and approximately 10% of those with primary sclerosing cholangitis had a bone density value below the fracture threshold, osteoporosis generally being associated with advanced disease [26]. In a more recent study, no increase was found in the incidence of metabolic bone disease in primary biliary cirrhosis patients who had been treated with regular calcium and vitamin D supplements [27].
Table 11.2. Factors increasing risk of bone disease in chronic cholestasis
| General | Reduced physical activity |
| Low body mass index | |
| Increasing age | |
| Female sex | |
| Reduced sunlight exposure | |
| Cholestasis | Vitamin D and K deficiency |
| Reduced calcium availability | |
| Increased serum bilirubin | |
| Genetic | Vitamin D receptor genotype |
| Hormonal | Menopause/ hypogonadism |
| Steroid therapy |
Bone disease manifests as loss of height, back pain (usually midthoracic or lumbar), collapsed vertebrae and fractures with minimal trauma, particularly of ribs. Spinal X-rays may show vertebrae of low density, as well as compression. Bone mineral density may be measured by dual photon absorptiometry. The cause of osteoporosis in chronic cholestatic liver disease is multifactorial. Factors that may play a role include vitamin D, calcitonin, parathyroid hormone, growth hormone and sex steroids. External influences in cholestatic patients include immobility, poor nutrition and reduced muscle mass. Vitamin D levels may be reduced due to malabsorption, inadequate diet and reduced exposure to the sun. Activation of vitamin D, by 25-hydroxylation in liver and 1-hydroxylation in the kidney, is normal. In patients with primary biliary cirrhosis, polymorphisms of the vitamin D receptor (VDR) gene correlate with the degree of osteoporosis and vertebral fracture [28].
Early experience with liver transplantation showed that improved bone density was delayed until 1–5 years after transplant. Before recovery, spontaneous bone fractures were common, occurring in 35% of patients with primary biliary cirrhosis in the first year. Heavy exposure to corticosteroids for immunosuppression probably played a part in this increased fracture rate. Vitamin D levels may not return to normal for several months after transplantation and supplementation has been recommended [29].
It is important to consider vitamin D deficiency in patients with chronic cholestasis. Cholestatic patients may fail to go out in the sun or take an adequate diet. Absorption is poor due to steatorrhoea. As chelation by cholestyramine may exacerbate the deficiency, vitamin D supplements, ursodeoxycholic acid and lipid-soluble medications such as digoxin should be taken in the evening to distance them from possible chelation by cholestyramine taken with breakfast. Vitamin D deficiency may be masked by secondary hyperparathyroidism, which will result in a compensatory mobilization of calcium from bones to normalize serum levels. Therefore blood testing for osteomalacia needs to be after an overnight fast when hypophosphataemia due to secondary hyperparathyroidism is most easily detected. Confirmation that vitamin D deficiency is the cause can be obtained by measurement of plasma vitamin D levels.
A rarer manifestation of bone disease is painful osteoarthropathy in the wrists and ankles [30], a non-specific complication of chronic liver disease.
Changes in Copper Metabolism
Approximately 80% of absorbed copper is normally excreted in bile and lost in faeces. In chronic cholestasis (as in primary biliary cirrhosis, biliary atresia or sclerosing cholangitis), hepatic copper levels may equal or exceed those found in Wilson’s disease. However, in cholestasis the retained copper is sequestered and not hepatotoxic. Kayser–Fleischer rings may be seen. Finding copper-associated protein on orcein staining of liver tissue supports a diagnosis of cholestasis.
Development of Hepatocellular Failure
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


