Urinary Stone Disease: Introduction
Urinary calculi are the third most common affliction of the urinary tract, exceeded only by urinary tract infections and pathologic conditions of the prostate. They are common in both animals and humans. The nomenclature associated with urinary stone disease arises from a variety of disciplines. Struvite stones, for example, composed of magnesium ammonium phosphate hexahydrate, are named in honor of H.C.G. von Struve (1772–1851), a Russian naturalist. Before the time of von Struve, the stones were referred to as guanite, because magnesium ammonium phosphate is prominent in bat droppings. Calcium oxalate dihydrate is frequently referred to as weddellite, because it was commonly found in floor samples collected from the Weddell Sea in Antarctica. The history of the nomenclature associated with urinary stone disease is as intriguing as that of the development of the interventional techniques used in their treatment.
Urinary stones have plagued humans since the earliest records of civilization. The etiology of stones remains speculative. If urinary constituents are similar from each kidney and if there is no evidence of obstruction, why do most stones present in a unilateral fashion? Why do not small stones pass uneventfully down the ureter early in their development? Why do some people form one large stone and others form multiple small calculi? There is much speculation concerning these and other questions.
Advances in the surgical treatment of urinary stones have outpaced our understanding of their etiology. As clinicians, we are concerned with an expedient diagnosis and efficient treatment. Equally important is a thorough metabolic evaluation directing appropriate medical therapy and lifestyle changes to help reduce recurrent stone disease. Without such follow-up and medical intervention, stone recurrence rates can be as high as 50% within 5 years. Uric acid calculi can recur even more frequently. Physicians look forward to gaining a better understanding of this multifactorial disease process in hopes of developing more effective prophylaxis.
Renal and Ureteral Stones
Mineralization in all biologic systems has a common theme in that the crystals and matrix are intertwined. Urinary stones are no exception; they are polycrystalline aggregates composed of varying amounts of crystalloid and organic matrix. Theories to explain urinary stone disease are incomplete.
Stone formation requires supersaturated urine. Supersaturation depends on urinary pH, ionic strength, solute concentration, and complexation. Urinary constituents may change dramatically during different physiologic states from a relatively acid urine in a first morning void to an alkaline tide noted after meals. Ionic strength is determined primarily by the relative concentration of monovalent ions. As ionic strength increases, the activity coefficient decreases. The activity coefficient reflects the availability of a particular ion.
The role of solute concentrations is clear: The greater the concentration of two ions, the more likely they are to precipitate. Low ion concentrations result in undersaturation and increased solubility. As ion concentrations increase, their activity product reaches a specific point termed the solubility product (Ksp). Concentrations above this point are metastable and are capable of initiating crystal growth and heterogeneous nucleation. As solutions become more concentrated, the activity product eventually reaches the formation product (Kfp). Supersaturation levels beyond this point are unstable, and spontaneous homogeneous nucleation may occur.
Multiplying two ion concentrations reveals the concentration product. The concentration products of most ions are greater than established solubility products. Other factors must play major roles in the development of urinary calculi, including complexation. Complexation influences the availability of specific ions. For instance, sodium complexes with oxalate and decreases its free ionic form, while sulfates can complex with calcium. Crystal formation is modified by a variety of other substances found in the urinary tract, including magnesium, citrate, pyrophosphate, and a variety of trace metals. These inhibitors may act at the active crystal growth sites or as inhibitors in solution (as with citrate).
The nucleation theory suggests that urinary stones originate from crystals or foreign bodies immersed in supersaturated urine. This theory is challenged by the same arguments that support it. Stones do not always form in patients who are hyperexcretors or who are at risk for dehydration. Additionally, up to a third of stone formers’ 24-hour urine collections are completely normal with respect to stone-forming ion concentrations.
The crystal inhibitor theory claims that calculi form due to the absence or low concentration of natural stone inhibitors including magnesium, citrate, pyrophosphate, and a variety of trace metals. This theory does not have absolute validity, since many people lacking such inhibitors may never form stones, and others with an abundance of inhibitors may, paradoxically, form them.
Stones are composed primarily of a crystalline component. Crystals of adequate size and transparency are easily identified under a polarizing microscope. X-ray diffraction is preferred to assess the geometry and architecture of calculi. A group of stones from the same geographic location or the same historical time period typically have crystalline constituents that are common.
Multiple steps are involved in crystal formation, including nucleation, growth, and aggregation. Nucleation initiates the stone process and may be induced by a variety of substances, including proteinaceous matrix, crystals, foreign bodies, and other particulate tissues. Heterogeneous nucleation (epitaxy), which requires less energy and may occur in less saturated urine, is a common theme in stone formation. It should be suspected whenever an oriented conglomerate is found. A crystal of one type thereby serves as a nidus for the nucleation of another type with a similar crystal lattice. This is frequently seen with uric acid crystals initiating calcium oxalate formation. It takes time for these early nidi to grow or aggregate to form a stone incapable of passing with ease through the urinary tract.
How these early crystalline structures are retained in the upper urinary tract without uneventful passage down the ureter is unknown. The theory of mass precipitation or intranephronic calculosis suggests that the distal tubules or collecting ducts, or both, become plugged with crystals, thereby establishing an environment of stasis, ripe for further stone growth. This explanation is unsatisfactory; tubules are conical in shape and enlarge as they enter the papilla (ducts of Bellini), thereby reducing the possibility of ductal obstruction. In addition, urine transit time from the glomerulus into the renal pelvis is only a few minutes, making crystal aggregation and growth within the uriniferous tubules unlikely.
The fixed particle theory postulates that formed crystals are somehow retained within cells or beneath tubular epithelium. Alexander Randall noted whitish-yellow precipitations of crystalline substances occurring on the tips of renal papillae as submucosal plaques. These plaques are associated with both the vasa recta and the urinary collecting ducts and grow deep within the papilla. The tips of the plaques can be appreciated during endoscopy of the upper urinary tract. Carr hypothesized that calculi form in obstructed lymphatics and then rupture into adjacent fornices of a calyx. Arguing against Carr’s theory are the grossly visible early stone elements in areas remote from fornices.
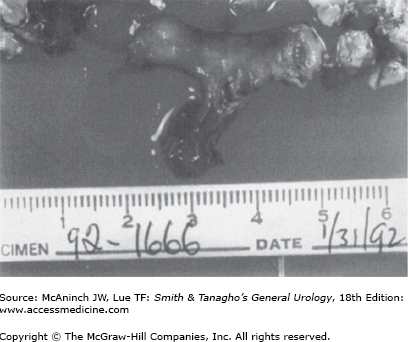
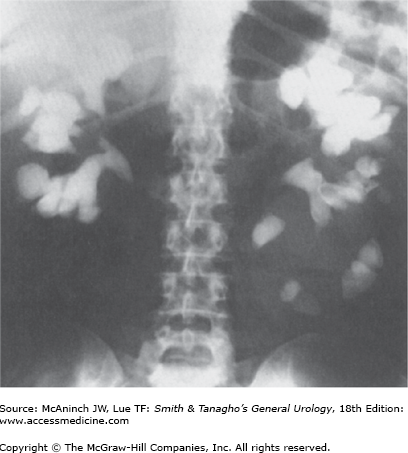
The amount of the noncrystalline, matrix component of urinary stones varies with stone type, commonly ranging from 2% to 10% by weight. It is composed predominantly of protein, with small amounts of hexose and hexosamine. An unusual type of stone called a matrix calculus can be associated with previous kidney surgery or chronic urinary tract infections and has a gelatinous texture (Figure 17–1). Histologic inspection reveals laminations with scant calcifications. On plain abdominal radiographs, matrix calculi are usually radiolucent and can be confused with other filling defects, including blood clots, upper-tract tumors, and fungal bezoars. Noncontrast computed tomography (CT) reveals calcifications and can help to confirm the diagnosis. The role of matrix in the initiation of ordinary urinary stones as well as matrix stones is unknown. It may serve as a nidus for crystal aggregation or as a naturally occurring glue to adhere small crystal components and thereby hinder uneventful passage down the urinary tract. Alternatively, matrix may have an inhibitory role in stone formation or may be an innocent bystander, playing no active role in stone formation.
Calcium is a major ion present in urinary crystals. The risk of urinary stones increases as fractional calcium absorption from the gut increases; calcium absorption from the gut decreases with increased calcium intake. Only 50% of plasma calcium is ionized and available for filtration at the glomerulus. More than 95% of the calcium filtered at the glomerulus is reabsorbed at both the proximal and distal tubules and limited amounts in the collecting tube. Less than 2% is excreted in the urine. Diuretic medications may exert a hypocalciuric effect by further decreasing calcium excretion. Many factors influence the availability of calcium in solution, including complexation with citrate, phosphate, and sulfate. An increase in monosodium urates and a decrease in urinary pH further interfere with this complexation and therefore promote crystal aggregation.
Oxalate is a normal waste product of metabolism and is relatively insoluble. Normally, approximately 10–15% of oxalate found in the urine originates from the diet; the vast majority is a metabolic by-product. Most of the oxalate that enters the large bowel is consumed by bacterial decomposition. Oxalobacter formigenes may alter the availability of oxalate absorption. Diet, however, can have an impact on the amount of oxalate found in the urine. Once absorbed from the small bowel, oxalate is not metabolized and is excreted almost exclusively by the proximal tubule. The presence of calcium within the bowel lumen is an important factor influencing the amount of oxalate that is absorbed. The control of oxalate in the urine plays a pivotal role in the formation of calcium oxalate calculi. Normal excretion ranges from 20 to 45 mg/day and does not change significantly with age. Excretion is higher during the day when one eats. Small changes in oxalate levels in the urine can have a dramatic impact on the supersaturation of calcium oxalate. The important precursors of oxalate are glycine and ascorbic acid; however, the impact of ingested vitamin C (<2 g/day) is negligible.
Hyperoxaluria may develop in patients with bowel disorders, particularly inflammatory bowel disease, small-bowel resection, and bowel bypass. Renal calculi develop in 5–10% of patients with these conditions. Chronic diarrhea with fatty stools results in a saponification process. Intraluminal calcium binds to the fat, thereby becoming unavailable to bind to oxalate. The unbound oxalate is readily absorbed from the gut and increases urinary oxalate levels.
Excessive oxalate may occur secondary to the accidental or deliberate ingestion of ethylene glycol (partial oxidation to oxalate). This may result in diffuse and massive deposition of calcium oxalate crystals and may occasionally lead to renal failure.
Phosphate is an important buffer and complexes with calcium in urine. It is a key component in calcium phosphate and magnesium ammonium phosphate stones. The excretion of urinary phosphate in normal adults is related to the amount of dietary phosphate (especially in meats, dairy products, and vegetables). The small amount of phosphate filtered by the glomerulus is predominantly reabsorbed in the proximal tubule. Parathyroid hormone inhibits this reabsorption. The predominant crystal found in patients with hyperparathyroidism is phosphate, in the form of hydroxyapatite, amorphous calcium phosphate, and carbonate apatite.
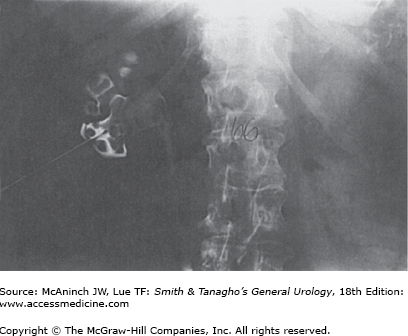
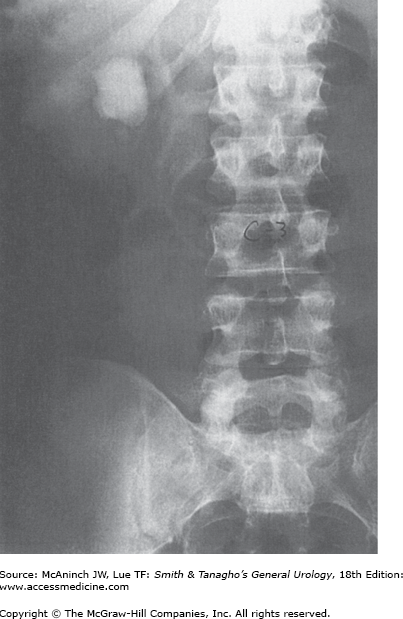
Uric acid is the by-product of purine metabolism. The pKa of uric acid is 5.75. Undissociated uric acid predominates with pH values less than this. Elevated pH values increase urate, which is soluble. Approximately 10% of the filtered uric acid finds its way into the urine. Other defects in purine metabolism may result in urinary stone disease. Rarely, a defect in xanthine dehydrogenase results in increased levels of xanthine; the xanthine may precipitate in urine, resulting in stone formation. Unusual alterations in adenine metabolism may result in the production of 2,8-dihydroxyadeninuria, which is poorly soluble in urine and may develop into a urinary stone. This results from a deficiency of adenine phosphoribosyltransferase (APRT). Pure uric acid crystals and calculi are relatively radiolucent and may not be identified on plain abdominal films (Figure 17–2). They are visible on noncontrast CT images and are suggestive when found to have low Hounsfield units (HU). Some uric acid calculi may be partially radiopaque, however, because of associated calcium phosphate deposits.
Although not identified as one of the major constituents of most urinary calculi, sodium plays an important role in regulating the crystallization of calcium salts in urine. Sodium is found in higher than expected concentrations in the core of renal calculi and may play a role in initiating crystal development and aggregation. High dietary sodium intake increases urinary calcium excretion and increases the urinary levels of monosodium urates that promote stone growth. This reduces the ability of urine to inhibit calcium oxalate crystal agglomeration. These effects are thought to be due to a sodium-induced increase in bicarbonaturia and decrease in serum bicarbonate. Interestingly, increased dietary sodium intake increases ones thirst and increases voided volumes. Conversely, a reduction in dietary sodium helps to reduce recurrent calcium nephrolithiasis.
Citrate is a key factor affecting the development of calcium urinary stones. A deficiency commonly is associated with stone formation in those with chronic diarrhea or renal tubular acidosis type I (distal tubular defect) and in patients prescribed chronic thiazide therapy. Citrate plays a pivotal role in the citric acid cycle in renal cells. Metabolic stimuli that consume this product (as with intracellular metabolic acidosis due to fasting, hypokalemia, or hypomagnesemia) reduce the urinary excretion of citrate. Estrogen increases citrate excretion and may be a factor that decreases the incidence of stones in women, especially during pregnancy. Alkalosis also increases citrate excretion.
Dietary magnesium deficiency is associated with an increased incidence of urinary stone disease. Magnesium is a component of struvite calculi. Experimentally, lack of dietary magnesium is associated with increased calcium oxalate stone formation and calcium oxalate crystalluria. The exact mechanism whereby magnesium exerts its effect is undefined. Dietary magnesium supplements typically do not protect against stone formation.
Urinary sulfates may help prevent urinary calculi. They can complex with calcium. These sulfates occur primarily as components of longer urinary proteins, such as chondroitin sulfate and heparin sulfate.
Inhibitors of urinary stone formation other than citrate, magnesium, and sulfates have been identified. These consist predominantly of urinary proteins and other macromolecules such as glycosaminoglycans, pyrophosphates, and uropontin. Although citrate appears to be the most active inhibitory component in urine, these substances demonstrate a substantial role in preventing urine crystal formation. The N-terminal amino acid sequence and the acidic amino acid content of these protein inhibitors, especially their high aspartic acid content, appear to play pivotal inhibitory roles. Fluoride may be an inhibitor of urinary stone formation.
Calcifications can occur and accumulate in the collecting system, resulting in nephrolithiasis. Eighty to eighty-five percent of all urinary stones are calcareous.
Calcium nephrolithiasis is most commonly due to elevated urinary calcium, elevated urinary uric acid, elevated urinary oxalate, or a decreased level of urinary citrate. Hypercalciuria is found as a solitary defect in 12% of patients and in combination with other defects in an additional 18%. Hyperuricosuria is identified as a solitary defect in 8% of patients and associated with additional defects in 16%. Elevated urinary oxalate is found as a solitary finding in 5% of patients and as a combined defect in 16%. Finally, decreased urinary citrate is found as an isolated defect in 17% of patients and as a combined defect in an additional 10%. Approximately one-third of patients undergoing a full metabolic evaluation will find no identifiable metabolic defect.
Symptoms from stones are secondary to obstruction, with resultant pain, infection, nausea, and vomiting, and rarely culminate in renal failure. Asymptomatic hematuria or repetitive urinary tract infections recalcitrant to apparently appropriate antibiotics should lead one to suspect a possible urinary stone. Calcifications within the parenchyma of the kidney, known as nephrocalcinosis, rarely cause symptoms, however, and usually are not amenable to traditional therapies appropriate for urinary stone disease (Figure 17–3). Nephrocalcinosis is frequently encountered with renal tubular acidosis and hyperparathyroidism. Nephrolithiasis and nephrocalcinosis frequently coexist. Most patients with nephrolithiasis, however, do not have obvious nephrocalcinosis.
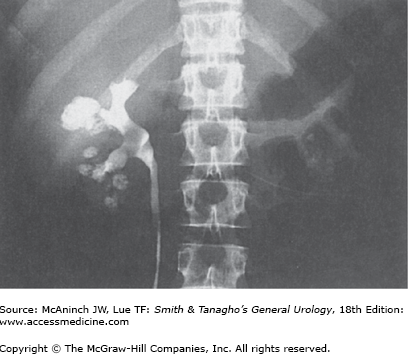
Nephrocalcinosis may result from a variety of pathologic states. Ectatic collecting tubules, as seen with medullary sponge kidney, are common; this is frequently a bilateral process. Theoretically, obstructed collecting ducts may be a source of chronic flank pain. Increased calcium absorption from the small bowel is common with sarcoidosis, milk-alkali syndrome, hyperparathyroidism, and excessive vitamin D intake. Disease processes resulting in bony destruction, including hyperparathyroidism, osteolytic lesions, and multiple myeloma, are a third mechanism. Finally, dystrophic calcifications forming on necrotic tissue may develop after a renal insult.
Normal calcium intake averages approximately 900–1000 mg/day. Approximately one-third is absorbed by the small bowel, and of that portion, approximately 150–200 mg is obligatorily excreted in the urine. A large reservoir of calcium remains in the bone. Most dietary calcium is excreted in the stool. Absorptive hypercalciuria is secondary to increased calcium absorption from the small bowel, predominantly from the jejunum. This results in an increased load of calcium filtered from the glomerulus. The result is suppression of parathyroid hormone, leading to decreased tubular reabsorption of calcium, culminating in hypercalciuria (>4 mg/kg). This physiologic cascade is in response to the primary defect, an increased absorption of calcium from the small bowel.
Absorptive hypercalciuria traditionally has been subdivided into three types. Type I absorptive hypercalciuria is independent of diet and represents 15% of all calcareous calculi. There is an elevated urinary calcium level (>150–200 mg/24 hour) even during a calcium-restricted diet. Cellulose phosphate is an effective nonabsorbable exchange resin. This effectively binds the calcium in the gut, preventing bowel absorption. Cellulose phosphate has no impact on the calcium transport defect. Urinary calcium excretion returns to normal values with therapy.
Cellulose phosphate must be taken with meals to be available when calcium is ingested. A typical dose is 10–15 g orally in three divided doses and is well tolerated. This therapy is relatively contraindicated in postmenopausal women and in children during their active growth cycles. Inappropriate use may lead to a negative calcium balance and a secondary hyperparathyroid state. As with all stone formers, long-term follow-up is required. Cellulose phosphate may bind other cations besides calcium, including magnesium. Secondary hyperoxaluria may develop owing to decreased calcium in the gut. See the section on hyperoxaluria for a more detailed discussion.
Hydrochlorothiazides are an alternative, more common treatment for type I absorptive hypercalciuria. Initially, there is a reduction in renal excretion of calcium. The increased absorbed calcium is likely deposited in bone. Eventually, the bone reservoir reaches its capacity and the drug becomes less effective. Hydrochlorothiazides have limited long-term efficacy (approximately 3–5 years). These drugs have no effect on the defective bowel transport system. Hydrochlorothiazides may be alternated with cellulose phosphate as an effective treatment regimen.
Type II absorptive hypercalciuria is dietary dependent. There is no specific medical therapy. Calcium excretion returns to normal on a calcium-restricted diet. These are the rare patients that should limit their calcium intake to 400–600 mg/day. Type II absorptive hypercalciuria is not as severe as type I.
Type III absorptive hypercalciuria is secondary to a phosphate renal leak and accounts for 5% of all urinary calculi. Decreased serum phosphate leads to an increase in 1,25-dihydroxyvitamin D synthesis. The physiologic cascade culminates in an increased absorption of phosphate, and calcium, from the small bowel and an increased renal excretion of calcium—hence its classification as absorptive hypercalciuria. Successful treatment replaces bioavailable phosphate. Orthophosphate (Neutra-Phos, now available over the counter) inhibits vitamin D synthesis and is best taken as 250 mg three to four times daily. It is best taken after meals and before bedtime. Orthophosphates do not alter intestinal calcium absorption.
A subset (<10%) of patients with clinically obvious primary hyperparathyroidism present with nephrolithiasis. This group represents less than 5% of all patients with urinary stones and are more commonly seen in women. Patients with calcium phosphate stones, women with recurrent calcium stones, and those with both nephrocalcinosis and nephrolithiasis should be suspected of having hyperparathyroidism. Hypercalcemia and an elevated serum parathyroid level are the most consistent signs of hyperparathyroidism.
Parathyroid hormone results in a cascade of events starting with an increase in urinary phosphorus and a decrease in plasma phosphorus, followed by an increase in plasma calcium and a decrease in urinary calcium. Its action on the kidney and on the bone is independent of each other. Ultimately, renal damage is secondary to the hypercalcemia. It limits the concentrating ability of the kidney and impairs the kidney’s ability to acidify urine. Surgical removal of the offending parathyroid adenoma is the most effective way of treating this disease. However, up to 25% of these patients will develop new urinary stones after successful surgery, most commonly seen in men. Attempts at long-term medical management are challenging.
Hypercalciuria of renal origin is due to an intrinsic renal tubular defect in calcium excretion. This creates a physiologically vicious cycle. Excessive urinary calcium excretion results in a relative decrease in serum calcium, which leads to a secondarily increased parathyroid hormone level that mobilizes calcium from the bone and increases calcium absorption from the gut. This step completes the pathologic cycle by delivering increased levels of calcium back to the kidney, whereby the renal tubules excrete large amounts of calcium. These patients have an elevated fasting urinary calcium level, normal serum calcium level, and a secondarily elevated parathyroid hormone level.
Renal hypercalciuria is effectively treated with hydrochlorothiazides. Unlike their role in type I absorptive hypercalciuria, in this setting hydrochlorothiazides have a durable long-term effect. As a diuretic, they decrease the circulating blood volume and subsequently stimulate proximal tubular absorption of calcium as well as other constituents. They also increase reabsorption at the distal tubule. Both mechanisms correct the secondary hyperparathyroid state.
Hypercalciuric states may result in elevated parathyroid levels. To help differentiate primary hyperparathyroidism from secondary hyperparathyroidism in patients with urinary stone disease, one can prescribe a hydrochlorothiazide challenge (50 mg twice a day for 7–10 days). Patients with secondary hyperparathyroidism will have a return to normal serum parathyroid levels, while those with primary hyperparathyroidism will continue to have elevated serum values.
Hyperuricosuric calcium nephrolithiasis is due to either an excessive dietary intake of purines or an increase in endogenous uric acid production. In both situations, there is an increase in urinary monosodium urates. Monosodium urates absorb and adsorb urinary stone inhibitors and facilitate heterogeneous nucleation.
Patients have elevated urinary uric acid levels (>600 mg/24 hour in women and >750 mg/24 hour in men) and consistently have a urinary pH >5.5. The urinary pH helps to differentiate hyperuricosuric calcium from hyperuricosuric uric acid stone formation.
Patients with excessive purine oral intake can be successfully treated by changing to a low purine diet. Those with excessive endogenous uric acid production can be treated with allopurinol. Allopurinol is a xanthine oxidase inhibitor and reduces uric acid synthesis and renal excretion of uric acid. It also inhibits uric acid–calcium oxalate crystallization. Allopurinol has many potential side effects, including a variety of skin rashes and, rarely, liver toxicity. Potassium citrate is an alternative treatment, especially when associated with hypocitraturia.
Hyperoxaluric calcium nephrolithiasis is secondary to increased urinary oxalate levels (>40 mg/24 hour). It is frequently found in patients with inflammatory bowel disease or other chronic diarrheal states that result in severe dehydration. It is rarely associated with excessive oxalate intake, as seen in poisoning with ethylene glycol or endogenous overproduction.
Chronic diarrheal states alter oxalate metabolism. Malabsorption leads to increased luminal fat and bile salts. Intraluminal gut calcium readily and preferentially binds to fat and bile resulting in a saponification process. The intraluminal gut calcium that normally would have bound to oxalate is thus decreased. The unbound oxalate is readily absorbed and is unaffected by the usual metabolic inhibitors of energy-dependent pumps. Bile salts may increase the passive bowel absorption of oxalate. A small increase in oxalate absorption and subsequent urinary excretion dramatically increases the formation product of calcium oxalate. This increases the potential for heterogeneous nucleation and crystal growth in this metastable environment. All patients with increased urinary excretion of oxalate do not necessarily form calcium oxalate calculi, other factors must be contributory.
Enteric hyperoxaluric calcium nephrolithiasis is successfully treated with oral calcium supplementation. The calcium binds to the intraluminal oxalate and thus limits its absorption. It must be given with meals when the oxalate is present. Other oral cations are effective binders including magnesium supplements. An alternative therapy includes a diet limited to medium-chain fatty acids and triglycerides; however, it is poorly tolerated by patients. Equally difficult is an attempt to alter oxalate intake. Unless large amounts of specific oxalate-rich foods can be excluded, an alternative diet may result in increased oxalate levels.
Primary hyperoxaluria is a rare hereditary disease. It is associated with calcium oxalate renal calculi, nephrocalcinosis, and other distant deposits of oxalate, culminating in progressive renal failure and eventual death. Type I is associated with an enzyme deficiency of 2-oxoglutarate:glyoxylate carboligase, resulting in elevated urinary levels of glycolic acid and oxalic acid. Type II has increased excretory levels of l-glyceric acid rather than elevated levels of glycolic acid. It is associated with a d-glycerate dehydrogenase enzyme deficiency. This ultimately results in the accumulation of hydroxy pyruvate, which is eventually converted to oxalate. Oxalate crystal deposits develop rapidly in transplanted kidneys. Combined liver and renal transplantation has cured this previously fatal rare disease.
Citrate is an important inhibitor of urinary stone disease. Increased metabolic demands on the mitochondria of proximal renal tubular cells decrease the excretion of citrate. Such conditions include intracellular metabolic acidosis, hypokalemia (as with thiazide therapy), fasting, hypomagnesemia, androgens, gluconeogenesis, and an acid-ash diet. Citrate may be consumed in the urine by bacteria during a urinary tract infection. The cause of hypocitraturia may be unknown in some cases. In contrast, alkalosis, alkaline-ash diet, estrogen, and vitamin D increase urinary citrate levels.
Citrate acts in solution and complexes with calcium. This decreases the ionic calcium concentration and thus the activity product. Citrate decreases agglomeration, spontaneous nucleation, and crystal growth of calcium oxalate. It also decreases calcium oxalate calculi by decreasing monosodium urates that can absorb inhibitors and facilitate heterogeneous nucleation.
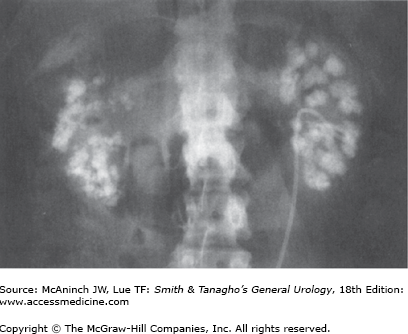
Hypocitraturic (<450 mg/24 h) calcium nephrolithiasis is associated commonly with renal tubular acidosis type I (distal tubule) (Figure 17–4), thiazide therapy (accompanied by potassium wastage), and chronic diarrhea. Treatment is successful with potassium citrate supplementation. Routine dosage is 20–30 mEq two to three times daily (tablet, crystal or liquid preparation) and is usually well tolerated. Six to eight glasses of lemonade can increase urinary citrate excretion by approximately 150 mg/24 h and thus either limit or eliminate the need for pharmacologic citrate supplementation.
Struvite stones are composed of magnesium, ammonium, and phosphate (MAP). They are found most commonly in women and may recur rapidly. They frequently present as renal staghorn configured calculi (branched with stones occupying the renal pelvis and at least to renal infundibuli) and rarely present as obstructing ureteral stones except after surgical intervention (Figure 17–5). Struvite stones are infection stones associated with urea-splitting organisms, including Proteus, Pseudomonas, Providencia, Klebsiella, Staphylococci, and Mycoplasma. The high ammonium concentration derived from the urea-splitting organisms results in an alkaline urinary pH. The urinary pH of a patient with a MAP stone rarely is <7.2 (normal urinary pH is 5.85). It is only at this elevated urinary pH (>7.19) that MAP crystals precipitate. MAP crystals are soluble in the normal urinary pH range of 5–7. Preoperative bladder cultures do not necessarily reflect the bacteriologic composition found in calculi. Foreign bodies and neurogenic bladders may predispose patients to urinary infections and subsequent struvite stone formation. Massive diuresis does not prevent struvite calculi. Women with recurrent non–Escherichia coli urinary infections despite apparently appropriate antibiotic therapy should be evaluated for struvite calculi with a conventional kidney–ureter–bladder (KUB) radiograph or renal ultrasound, or both. It is impossible to sterilize such calculi with antibiotics. Culture-specific antibiotics can reduce urease levels and help reduce stone recurrence. Stone removal is therapeutic.
Long-term management is optimized with the removal of all foreign bodies, including catheters of all varieties. A short ileal loop urinary diversion helps decrease the risk of stones in those with supravesical urinary diversion. All stone fragments should be removed with or without the aid of follow-up irrigations. Hemiacidrin (Renacidin) irrigations should be used with caution if at all. Rapid magnesium toxicity can result in death even with a low-pressure irrigation setup (<20 cm water pressure) and negative daily urine cultures. Acetohydroxamic acid inhibits the action of bacterial urease, thereby reducing the urinary pH and decreasing the likelihood of precipitation. Most patients have a difficult time tolerating this medication.
Uric acid stones comprise <5% of all urinary calculi and are usually found in men. Patients with gout, myeloproliferative diseases, or rapid weight loss and those treated for malignant conditions with cytotoxic drugs have an increased incidence of uric acid lithiasis. Most patients with uric acid calculi, however, do not have hyperuricemia. Elevated urinary uric acid levels are frequently due to dehydration and excessive purine intake. Patients present with a urinary pH consistently <5.5, in contrast to patients with hyperuricosuric calcium nephrolithiasis, who have a urinary pH >5.5. As the urinary pH increases above the dissociation constant pKa of 5.75, it dissociates into a relatively soluble urate ion. Treatment is centered on maintaining a urine volume >2 L/day and a urinary pH >6.0. Reducing dietary purines or the administration of allopurinol also helps reduce uric acid excretion. Alkalinization, however, is the mainstay of therapy (with oral sodium bicarbonate, potassium bicarbonate, potassium citrate, or intravenous 1/6 normal sodium lactate) and may dissolve calculi (the rate of dissolution is dependent on the stone surface area). Stone fragments after lithotripsy have a dramatically increased surface area compared with intact stones and thus will dissolve more rapidly. Dissolution proceeds at approximately 1 cm of stone (as seen on KUB) per month, with compliant alkalinization.
Cystine lithiasis is secondary to an inborn error of metabolism resulting in abnormal intestinal (small bowel) mucosal absorption and renal tubular absorption of dibasic amino acids, including cystine, ornithine, lysine, and arginine. The genetic defects associated with cystinuria have now been mapped to chromosome 2p.16 and more recently to 19q13.1. Cystine lithiasis is the only clinical manifestation of this defect. Classic cystinuria is inherited in an autosomal recessive fashion. Homozygous expression has a prevalence of 1:20,000, while the heterozygous expression is 1:2000. It represents 1–2% of all urinary stones, with a peak incidence in the second or third decade. Homozygous cystinurics excrete more than 250 mg/day, resulting in constant supersaturation. Heterozygous patients usually excrete 100–150 mg/day. Unaffected patients typically excrete <40 mg/day. Approximately 400 mg/L of cystine can remain in solution at a urinary pH of 7.0. As the urinary pH increases above 7.0, the amount of soluble cystine increases exponentially.
The solubility of cystine is pH dependent, with a pK of approximately 8.1. There is no difference in the solubility curves in normal versus patients with cystinuria. There is no known inhibitor for cystine calculi, and cystine stone formation is completely dependent on excessive cystine excretion. Cystine stones are frequently associated with calcium calculi and their related metabolic abnormalities. They may present as single, multiple, or staghorn configured stones. The diagnosis is suspected in patients with a family history of urinary stones and the radiographic appearance of a faintly opaque, ground-glass, smooth-edged stone (Figure 17–6). Urinalysis frequently reveals hexagonal crystals. The stones have an amber color. Stone analysis confirms the diagnosis. Quantitative urinary cystine evaluation helps confirm the diagnosis and differentiate heterozygous from homozygous states. It is also important to help titrate medical therapy.
Medical therapy includes high fluid intake (>3 L/d, day and night) and urinary alkalinization. Patients should monitor their pH with Nitrazine indicator paper and keep their pH values above 7.5. It is difficult or impossible to maintain levels >8.0. A low-methionine (precursor to cystine) diet has limited impact, as most of the cystine is endogenous and most of the ingested methionine is incorporated into protein. Glutamine, ascorbic acid, and captopril are effective in some patients. Penicillamine can reduce urinary cystine levels. It complexes with the amino acid, and this complex is dramatically more soluble. Treatment should be titrated with quantitative urinary cystine values. Many patients poorly tolerate penicillamine, reporting skin rashes (discrete or confluent macules with occasional itching), loss of taste, nausea, vomiting, and anorexia. It may inhibit pyridoxine, which should be supplemented during treatment (50 mg/d). Mercaptopropionylglycine (Thiola), 300–1200 mg in divided doses, with initial dosing matched with total quantitative cystine excretion (mg per mg) forms a soluble complex with cystine and can reduce stone formation and is the most commonly used sulfide-binding drug. Side effects and frequent dosing, however, decrease patient compliance.
Surgical treatment is similar to that for other stones except that most stones are recalcitrant to extracorporeal shock wave lithotripsy (SWL). One should have a low threshold to proceed with percutaneous stone extraction in symptomatic patients. Despite optimum medical therapy, a high stone recurrence rate frequently frustrates both patient and physician. Minimally invasive techniques and optimum medical therapy are paramount.
Xanthine stones are secondary to a congenital deficiency of xanthine dehydrogenase. This enzyme normally catalyzes the oxidation of hypoxanthine to xanthine and of xanthine to uric acid. It is of interest that allopurinol, used to treat hyperuricosuric calcium nephrolithiasis and uric acid lithiasis, produces iatrogenic xanthinuria. Blood and urine levels of uric acid are lowered, and hypoxanthine and xanthine levels are increased; however, there are no case reports of xanthine stone formation resulting from allopurinol treatment. It is unlikely that allopurinol completely inhibits xanthine dehydrogenase. Urinary stones develop in approximately 25% of patients with this enzymatic deficiency. The stones are radiolucent and are tannish yellow in color. Treatment should be directed by symptoms and evidence of renal obstruction. High fluid intake and urinary alkalinization are required for prophylaxis. If stones recur, a trial of allopurinol and a purine-restricted diet is appropriate.
Protease inhibitors are a popular and effective treatment in patients with acquired immunodeficiency syndrome. Indinavir is the most common protease inhibitor that results in radiolucent stones in up to 6% of patients who are prescribed this medication. Indinavir calculi are the only urinary stones to be radiolucent on noncontrast CT scans. They may be associated with calcium components and in these situations will be visible on noncontrast CT images. Temporary cessation of the medication with intravenous hydration frequently allows these stones to pass. The stones are tannish red and usually fall apart during basket extraction.
Silicate stones are very rare and are usually associated with long-term use of antacids containing silica. Surgical treatment is similar to that of other calculi.
Triamterene stones are radiolucent and have been identified with an increased frequency. They are associated with antihypertensive medications containing triamterene, such as Dyazide. Discontinuing the medication eliminates stone recurrences. Other medications that may become stone constituents include glafenine and antrafenine.
Rarely, patients arrive at an emergency room at an odd hour feigning signs and symptoms of passing a urinary stone in hopes of obtaining pain medications. They may add blood to their urine and give a believable story of a severe allergy to intravenous contrast medium. Occasionally, patients present a fake urinary stone, with specks of paint or other obvious curiosities. Such patients have Munchausen syndrome, and the diagnosis is difficult and made by exclusion.
Upper-tract urinary stones frequently cause pain when passing down the ureter. The character of the pain depends on the location. Calculi small enough to venture down the ureter usually have difficulty passing through the ureteropelvic junction, or entering the bladder at the ureterovesical junction (Figure 17–7). More than 60% of patients presenting with ureteral colic will have stones within 3 cm of the ureterovesical junction.
Figure 17–7.
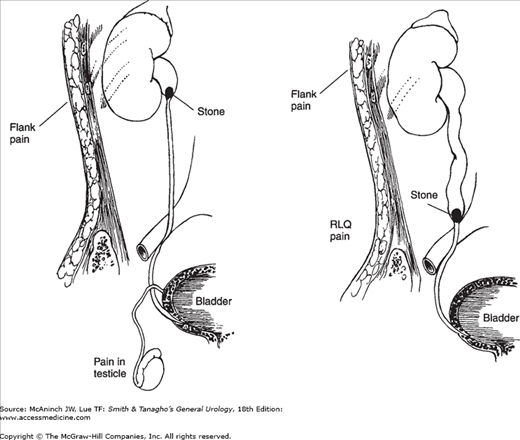
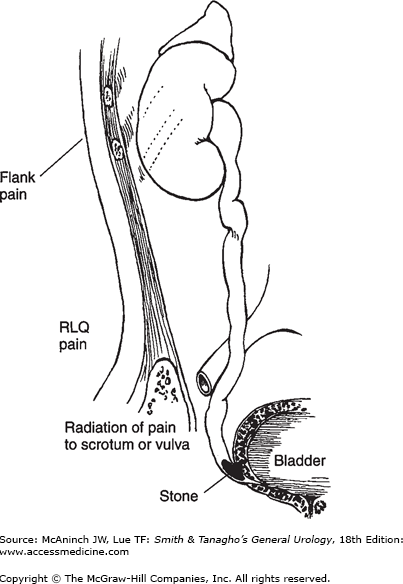
Radiation of pain with various types of ureteral stone. Upper left: Ureteropelvic stone. Severe costovertebral angle pain from capsular and pelvic distention; acute renal and urethral pain from hyperperistalsis of smooth muscle of calyces, pelvis, and ureter, with pain radiating along the course of the ureter (and into the testicle, since the nerve supply to the kidney and testis is the same). The testis is hypersensitive. Upper right: Mid ureteral stone. Same as described earlier but with more pain in the lower abdominal quadrant. Left: Low ureteral stone. Same as described earlier, with pain radiating into bladder, vulva, or scrotum. The scrotal wall is hyperesthetic. Testicular sensitivity is absent. When the stone approaches the bladder, urgency and frequency with burning on urination develop as a result of inflammation of the bladder wall around the ureteral orifice.
Renal colic and noncolicky renal pain are the two types of pain originating from the kidney. Renal colic usually is caused by stretching of the collecting system or ureter, whereas noncolicky renal pain is caused by distention of the renal capsule. These symptoms may overlap, making a clinical differentiation difficult or impossible. Urinary obstruction is the main mechanism responsible for renal colic. This may be mimicked by the pain a patient experiences when a retrograde ureteropyelogram is performed under local anesthesia, with excessive pressure resulting in overdistention of the collecting system. This pain is due to a direct increase in intraluminal pressure, stretching nerve endings.
Renal colic does not always wax and wane or come in waves like intestinal or biliary colic but may be relatively constant. Renal colic implies an intraluminal origin. Patients with renal calculi have pain primarily due to urinary obstruction.
Local mechanisms such as inflammation, edema, hyperperistalsis, and mucosal irritation may contribute to the perception of pain in patients with renal calculi. In the ureter, however, local pain is referred to the distribution of the ilioinguinal nerve and the genital branch of the genitofemoral nerve, whereas pain from obstruction is referred to the same areas as for collecting system calculi (flank and costovertebral angle), thereby allowing discrimination.
The vast majority of urinary stones present with the acute onset of pain due to acute obstruction and distention of the upper urinary tract. The severity and location of the pain can vary from patient to patient due to stone size, stone location, degree of obstruction, acuity of obstruction, and variation in individual anatomy (eg, intrarenal vs extrarenal pelvis). The stone burden does not correlate with the severity of the symptoms. Small ureteral stones frequently present with severe pain, whereas large staghorn configured calculi may present with a dull ache or flank discomfort.
The pain frequently is abrupt in onset and severe, and may awaken a patient from sleep. The severity of the pain is worsened by the unexpected nature of its onset. Patients frequently move constantly into unusual positions in an attempt to relieve the pain. This movement is in contrast to the lack of movement of someone with peritoneal signs; such a patient lies in a stationary position.
The symptoms of acute renal colic depend on the location of the calculus; several regions may be involved: renal calyx, renal pelvis, upper and mid ureter, and distal ureter. An orderly progression of symptoms as a stone moves down the urinary tract is the exception.
Stones or other objects in calyces or caliceal diverticula may cause obstruction and renal colic. In general, nonobstructing stones cause pain only periodically, owing to intermittent obstruction. The pain is a deep, dull ache in the flank or back that can vary in intensity from severe to mild. The pain may be exacerbated after consumption of large amounts of fluid. Radiographic imaging may not reveal evidence of obstruction despite the patient’s complaints of intermittent symptoms. It remains unclear how much of this pain is related to local mucosal irritation with activation of chemoreceptors. The presence of infection or inflammation in the calyx or diverticulum (eg, milk of calcium) in addition to obstruction may contribute to pain perception. Caliceal calculi occasionally result in spontaneous perforation with urinoma, fistula, or abscess formation.
Caliceal calculi are frequently small and numerous and appear to be able to pass spontaneously. Long-term retention against the flow of urine and against the forces of gravity and antegrade peristalsis suggests a significant element of obstruction. Effective long-term treatment requires stone extraction and elimination of the obstructive component.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


