Fig. 26.1
Production and metabolism of uric acid. XO xanthine oxidase, XDH xanthine dehydrogenase, NAD nicotinamide adenine dinucleotide
The best known consequence of an elevated uric acid in humans is gout. Because of the limited solubility of uric acid, gout is characterized by the deposition of urate crystals in synovial joints, occasionally with tophi formation. However, there are also a number of well-known renal and cardiovascular manifestations associated with hyperuricemia. There are three major different types of uric acid-induced renal disease: acute uric acid nephropathy, chronic uric acid nephropathy, and uric acid nephrolithiasis. In the following chapter, the major associations of uric acid with renal disease will be reviewed. Newer data will be presented regarding the role of uric acid in the progression of other renal diseases and in mediating intrarenal vascular disease and hypertension. Clinical implications and treatment strategies of asymptomatic hyperuricemia will be also discussed.
Uric Acid Metabolism and Homeostasis
Production of Uric Acid
Uric acid is a weak acid trioxypurine (M.W. 168) that is composed of a pyrimidine and imidazole substructure with oxygen molecules, which is produced from metabolic conversion of either dietary or endogenous purines, primarily in the liver, muscle, and intestine [1]. The immediate precursor of uric acid is xanthine, which is degraded into uric acid by xanthine oxidoreductase. Xanthine oxidoreductase may attain two interconvertible forms, namely, xanthine dehydrogenase or xanthine oxidase [4]. Most xanthine oxidoreductase is in the xanthine dehydrogenase form in vivo [4, 5], which is transformed into xanthine oxidase by irreversible proteolytic cleavage or reversible oxidation in specific environment [6] including hypoxia. Xanthine oxidase uses molecular oxygen as electron acceptor and generates superoxide anion and other reactive oxygen species as by-products in the process of uric acid degradation, whereas xanthine dehydrogenase generates the reduced form of nicotinamide adenine dinucleotide (Fig. 26.1). Both exogenous (present in fatty meat, organ meats, and seafood) and endogenous purines are major sources of uric acid in humans. Approximately two-thirds of total body urate is produced endogenously, while the remaining one-third is accounted for by dietary purines. Purine-rich foods include beer and other alcoholic beverages, anchovies, sardines in oil, fish roes, herring, organ meat (liver, kidneys, sweetbreads), legumes (dried beans, peas), meat extracts, consomme, gravies, mushrooms, spinach, asparagus, and cauliflower [7].
Excretion of Uric Acid in Humans
The primary site of excretion of uric acid is the kidney. The normal urinary urate excretion is in the range of 250–750 mg/day, approximately 70 % of the daily urate production [8]. Although urate (the form of uric acid at blood pH of 7.4) is freely filtered in the glomerulus, there is evidence that both reabsorption and secretion occur in the proximal tubule and as a consequence the fractional urate excretion is only 8–10 % in the normal adult. Some adaptation occurs with renal disease, in which the fractional excretion of urate will increase to the 10–20 %. The remainder of uric acid excretion occurs through the gut, where uric acid is degraded by uricolytic bacteria. The gastrointestinal tract may eliminate up to one-third of the daily uric acid load in the setting of renal failure [9, 10].
The classic paradigm of uric acid excretion consists of a four-step model with glomerular filtration, reabsorption, secretion, and postsecretory reabsorption; the latter three processes occur in the proximal convoluted tubule [11, 12]. Ideas of the handling of uric acid by the kidney have changed greatly over the past few decades, with the identification, characterization, and isolation of transporters and channels mainly or exclusively restricted to urate transport [13–15]. Membrane vesicle studies have suggested the existence of two major mechanisms modulating urate reabsorption and secretion, consisting of a voltage-sensitive pathway and a urate/organic anion exchanger. Recently several of these transporters/channels have been identified (Fig. 26.2).
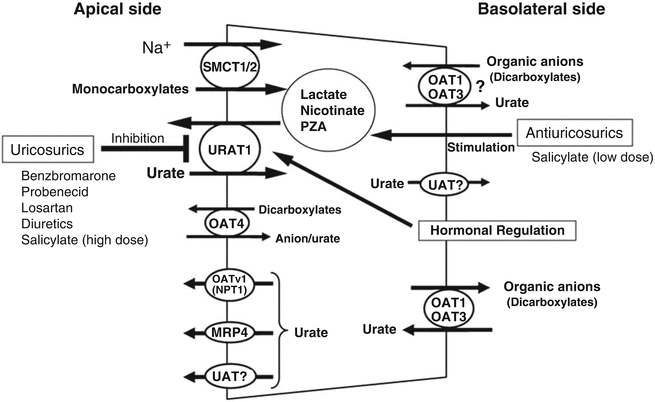
Fig. 26.2
Models of transcellular urate transport in the proximal tubule. From Anzai et al. [15]; used with permission
Uric acid transporter-1 (URAT-1), a member of the organic anion transporter (OAT) family, is identified as the major organic anion exchanger for uric acid on the apical (luminal brush border) side of the proximal tubular cell [13]. In the human kidney, urate is transported via URAT-1 across the apical membrane of proximal tubular cells, in exchange for anions being transported back into the tubular lumen to maintain electrical balance. Urate then moves across the basolateral membrane into the blood by other OATs, most likely OAT-1 and OAT-3 [16, 17]. URAT-1 has a high affinity for urate in exchange for lactate, ketones, α-ketoglutarate, and related compounds. Pyrazinamide, probenecid, losartan, and benzbromarone all inhibit urate uptake in exchange for chloride at the luminal side of the cell by competition with the urate exchanger. In humans, urate secretion is relatively minimal and URAT-1 is thought to be the major mechanism for regulating blood level of uric acid.
OAT4 is one of the apical urate transporters in the proximal tubule which has recently been identified as an organic anion–decarboxylate exchanger. OAT4 may be also a target of the uricosuric agent probenecid [18, 19].
Urate secretion may be mediated principally by a voltage-sensitive transporter, which is expressed ubiquitously and localizes to the apical side of the proximal tubule in the kidney. This may be the role of the urate transporter (UAT), which is in the galectin family [14]. UAT/galectin 9 is thought to be a housekeeping urate channel that serves in the efflux of urate produced by intracellular purine metabolism [20]. The role of UAT/galectin 9 as a urate transporter/channel in the apical membrane of proximal tubules remains to be elucidated.
Recently, a novel human renal apical organic anion efflux transporter, called MRP4, has been identified [21]. MRP4 is a member of the ATP-binding cassette transporter family. It may mediate the secretion of urate and other organic anions such as cAMP, cGMP, and methotrexate across the apical membrane of human renal proximal tubular cells. Human MRP4 is an ATP-dependent unidirectional efflux pump for urate with multiple allosteric substrate-binding sites [22].
Another protein involved in renal transport of urate is Tamm–Horsfall protein (THP), also known as uromodulin. THP is exclusively expressed and secreted by epithelial cells of the thick ascending limb and co-localizes with the Na, K, 2Cl transporter in lipid rafts in the apical cell membrane, suggesting a functional interaction [23]. Mutations in the human uromodulin gene have been identified in subjects with medullary cystic kidney disease type 2 and in patients with familial juvenile hyperuricemic nephropathy (FJHN) (see Section “Familial Juvenile Hyperuricemic Nephropathy”) [24, 25]. It is not yet known how the THP mutation leads to hyperuricemia, as most evidence suggests that uric acid handling is restricted to the proximal tubule. There is some evidence, however, that some urate secretion in the rat can occur distal to the proximal tubule [26]. Furthermore, there is also an evidence that the THP mutation may lead to sodium and water wasting and possibly stimulate urate reabsorption proximally [27].
Another novel insight regarding renal urate transport is a potential link between sodium and urate reabsortion as in a cotransport process [28]. A model of indirect coupling of sodium transport via SMCT1/2 and urate transport via URAT-1 with transcellular urate transport in the proximal tubule is shown in Fig. 26.2.
Uric Acid-Induced Nephropathy
Chronic Uric Acid Nephropathy
In patients who have had gout for many years, renal disease is common. Natural history studies prior to the availability of uric acid-lowering drugs reported that up to 25 % of gouty subjects developed proteinuria, 50 % developed renal insufficiency, and 10–25 % developed end-stage renal disease [29, 30]. Renal histologic changes are particularly common and have been observed in autopsy studies of 75–99 % of patients with gout. The classic definition of chronic uric acid nephropathy (also known as chronic urate nephropathy or gout nephropathy) is a form of chronic kidney disease (CKD) induced by the deposition of monosodium urate crystals in the distal collecting duct and the medullary interstitium with a secondary inflammatory reaction. The other primary histologic findings consist of arteriolosclerosis, glomerulosclerosis, and tubulointerstitial fibrosis. The exact pathophysiological mechanism for renal tophi formation is not clear since hyperuricemia and simple oversaturation of the urine is not a prerequisite [31].
This characteristic pathologic pattern has historically been termed “gouty nephropathy” and was thought to be a major etiology of chronic renal disease in the past [32]. Studies performed primarily in the 1970s and 1980s challenged the concept that gouty nephropathy was a true entity. Uric acid crystal deposition is usually focal and this does not adequately explain the diffuse pattern of the disease. Moreover, uric crystal deposition could be found in some autopsies where no renal disease was present [33]. One should also consider that most of the gouty subjects who had or developed renal disease had concomitant hypertension or were elderly as both conditions are associated with the development of microvascular disease, glomerulosclerosis, and tubulointerstitial fibrosis [34–36]. Yü and Berger [34–36] argued that, although gout was often associated with renal disease, mechanisms directly related to uric acid seem not to be responsible for renal pathology. For example, when they excluded gouty subjects with hypertension or who were older, they found little evidence that gout could cause renal disease.
Importantly, the effect of uric acid-lowering therapy on renal function in gout remains debatable. Although some studies suggested that lowering uric acid could improve the renal disease in gout [37, 38], other studies could not demonstrate any significant improvement [39]. Even in studies in which an improvement in renal disease was observed, the question remains whether better uric acid control consequently resulted in less use of nonsteroidal agents, which are considered to be nephrotoxic [40]. Finally, it became evident that some subjects with high uric acid and renal disease had other concomitant disorders such as lead nephropathy [41] or hereditary familial hyperuricemic nephropathy [42]. Although some investigations suggested that markedly elevated uric acid levels might cause chronic renal disease (usually at levels >13 mg/dL in men or >10 mg/dL in women) [43], most authorities concluded that uric acid was unlikely to cause a chronic nephropathy directly. Therefore, many authorities considered the term “gouty nephropathy” a misnomer [31, 44].
New Insights into Chronic Uric Acid Nephropathy
Despite skepticism about the clinical implication of chronic uric acid nephropathy, renewed interest in the role of gout and asymptomatic hyperuricemia has emerged in the last several years. First, it was recognized that there were questionable inherent assumptions in previous investigations [45]. For example, one implicit assumption was that primary pathogenesis of chronic uric acid nephropathy had to be uric acid crystal deposition with an exclusion of the possibility of other effects of hyperuricemia as a mediator of renal disease. It may also be inappropriate to use the presence of hypertension to explain every case of renal insufficiency in the gouty patient, since most subjects with essential hypertension have relatively preserved renal function. Furthermore, the analysis also assumed that the presence of hypertension was a separate cause of renal disease and that it had to be independent of the uric acid. These ideas led to a proposal to reinvestigate the role of uric acid in chronic renal disease.
Subsequently, a number of studies using multivariable regression modeling have examined the relationship between serum uric acid levels and the subsequent risk for developing chronic renal disease. Interestingly, several studies of the general population found that hyperuricemia was a strong independent risk factor for developing renal insufficiency. A serum uric acid of >7 mg/dL in men and >6 mg/dL in women confers a 10.8-fold increased risk in women and a 3.8-fold increased risk in men compared to those with normal uric acid levels [46]. This increased relative risk was independent of age, body mass index, systolic blood pressure, total cholesterol, serum albumin, glucose, smoking, alcohol use, exercise habits, proteinuria, and presence of hematuria. Indeed, an elevated uric acid was more predictive for the development of renal insufficiency than proteinuria. An elevated uric acid was also independently associated with a markedly increased risk of renal failure in another study of more than 49,000 male railroad workers [47]. Additional evidence that hyperuricemia may have a role in kidney disease was provided by a recent randomized, controlled clinical trial in which allopurinol therapy resulted in a slowing of renal disease progression in subjects with hyperuricemia and renal insufficiency associated with a significant decrease in uric acid level [48].
Another major breakthrough in understanding the role of uric acid on the progression of kidney disease came from experimental studies in which chronic mild hyperuricemia was induced in rats [49, 50]. Because rats have functional uricase, mild hyperuricemia can be induced by administering low concentrations of the uricase inhibitor, oxonic acid, to the diet [49, 50]. This resulted in serum uric acid levels that were only 1.5- to 3.0-fold greater than normal, and importantly this did not result in intratubular or interstitial urate crystal deposition. Over time, however, rats developed hypertension and progressive renal disease. Early in the course, the rats developed arteriolar thickening and rarely hyalinosis of the preglomerular arterioles, often accompanied by glomerular hypertrophy [50, 51]. Over time proteinuria developed and was followed by worsening vascular disease, glomerulosclerosis, and interstitial fibrosis [50]. The lesion was identical to that observed with nephrosclerosis of hypertension, with aging-associated glomerulosclerosis, and with gouty nephropathy, except for the absence of crystal deposition that had been observed in the latter condition. These findings raised the possibility that chronic hyperuricemia may cause renal disease and hypertension via a crystal-independent pathway.
Clinical Manifestations and Diagnosis of Chronic Uric Acid Nephropathy
Although histologic evidence of renal disease is almost invariably present, most subjects with long-standing gout will have either normal plasma creatinine or only mild renal insufficiency [34–36]. Renal blood flow is usually disproportionately low for the degree of renal insufficiency [34–36]. Fractional excretion of uric acid is usually less than 10 %. Proteinuria occurs in the minority of cases and, when present, is usually in the non-nephrotic range. The urinary sediment is also usually benign. However, hypertension is frequent, occurring in 50–60 % of subjects and increasing in prevalence as renal function worsens.
The diagnosis of chronic uric acid nephropathy is classically defined by histology, demonstrating chronic glomerulosclerosis, tubulointerstitial fibrosis, renal microvascular disease, and focal medullary crystal deposition. Chronic hyperuricemia can lead to precipitation of uric acid crystals (tophus formation) mainly in the distal collecting ducts and the interstitium. The crystal evokes a foreign body reaction with central accumulation of crystalloid material (monosodium urate) surrounded by a rim of inflammatory cells (Fig. 26.3). Recent experimental studies, however, suggest that crystal deposition is not necessary. Therefore, the diagnosis may become problematic because the other histologic findings are indistinguishable from benign nephrosclerosis (renal injury from essential hypertension) or with aging-associated renal disease. A disproportionately elevated serum uric acid in relation to the degree of renal insufficiency (such as a uric acid level of >9 mg/dL for a serum creatinine of <1.5 mg/dL, a uric acid of 10 mg/dL for a serum creatinine of 1.5–2.0 mg/dL, and a serum uric acid of >12 mg/dL when serum creatinine is >2.0 mg/dL) should make one consider the diagnosis of chronic uric acid nephropathy.
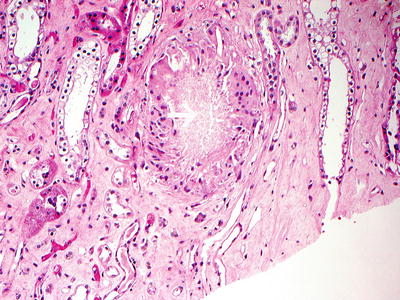
Fig. 26.3
Uric acid crystals (white arrow) surrounded by macrophages including giant cells (H and E ×40)
Management of Chronic Uric Acid (Gouty) Nephropathy
Although no specific studies have addressed management of chronic uric acid nephropathy, it is reasonable to treat gouty nephropathy similar to chronic renal disease of other etiologies. Therefore, one should achieve good blood pressure control (targeting a blood pressure of <140/90 mm Hg in nonproteinuric patients and 125/75 mm Hg in proteinuric patients). Given the known benefit of blocking the renin–angiotensin system on other renal diseases, and the observation that renin–angiotensin system blockade also prevents the renal injury induced with experimental hyperuricemia [49–51], we would recommend the use of an angiotensin receptor blocker and/or an angiotensin-converting enzyme inhibitor as part of the clinical regimen. Furthermore, losartan may be a particularly good choice as it is also uricosuric and will lower uric acid levels by affecting the urate–anion exchange in the proximal tubule through a mechanism that is not shared with other angiotensin receptor blockers [52]. The use of thiazide or loop diuretics is discouraged, as these drugs have as a side effect of raising the uric acid, and recent studies suggest they provide less renal protection than other antihypertensive agents available [45]. If a diuretic is required, the use of amiloride or spironolactone could be attempted with a careful monitoring for the development of hyperkalemia. Some subjects will nevertheless require a thiazide to control blood pressure.
The role of hypouricemic therapy in chronic uric acid nephropathy still remains controversial, and no definitive studies have been performed to resolve this important issue. Based on the experimental studies, it does seem reasonable to lower serum uric acid in subjects with hyperuricemia, particularly in individuals with markedly elevated levels (>8 mg/dL). In the setting of renal insufficiency, most uricosuric agents are relatively ineffective, although some success with benziodarone has been reported [53]. Allopurinol is the most commonly used hypouricemic agent because of its ability to lower serum uric acid regardless of the cause of hyperuricemia and the convenient once daily dosing. Allopurinol reduces the conversion of xanthine to uric acid through inhibition of xanthine oxidase, which is the rate-limiting step. Approximately 20 % of patients experience side effects with allopurinol and up to 5 % discontinuing therapy [54]. The most serious side effect is a rare hypersensitivity syndrome, which results in fever, rash, eosinophilia, hepatitis, renal failure, and in some cases death. In addition, oxypurinol, the functional metabolite of allopurinol, builds up in the setting of renal insufficiency. Dosing guidelines for allopurinol according to creatinine clearance have been published in an attempt to reduce the risk of the hypersensitivity syndrome (Table 26.1) [55]. A promising drug that is currently undergoing clinical trials is febuxostat, a nonpurine xanthine oxidase inhibitor that appears to have minimal side effects. Febuxostat may be more effective than allopurinol and for which no dose adjustment in renal disease is needed [56]. In addition to pharmacologic therapies, patients should be instructed to reduce ingestion of foods that can raise uric acid, such as foods with high-purine content, ethanol, and fructose. Management of hyperuricemia in CKD of other etiologies will be discussed in detail in the following sections.
Creatinine clearance (mL/min) | Maintenance dose allopurinol |
|---|---|
0 | 100 mg every 3 days |
10 | 100 mg every 2 days |
20 | 100 mg/day |
40 | 150 mg/day |
60 | 200 mg/day |
80 | 250 mg/day |
100 | 350 mg/day |
120 | 350 mg/day |
140 | 400 mg/day |
Acute Uric Acid Nephropathy
Acute uric acid nephropathy is characterized by acute oliguric renal failure with a rapid rise in serum uric acid level, which is almost always associated with uric acid precipitation within the tubules. This is most often due to overproduction and overexcretion of uric acid in patients with hematologic malignancy [57]. Tumor cell necrosis (“tumor lysis syndrome”) leads to a brisk increase of purine metabolism, hyperuricemia, and characteristic hyperuricosuria. Uncommon causes of acute uric acid nephropathy include tissue catabolism due to seizures or treatment of solid tumors, primary overproduction of uric acid due to the rare syndrome of hypoxanthine–guanine phosphoribosyltransferase deficiency, or hyperuricosuria due to decreased uric acid reabsorption in the proximal tubule, as seen in the patients with a Fanconi-like syndrome.
Serum uric acid levels may increase to greater than 15 mg/dL, resulting in a marked increase in urinary urate excretion that exceeds its solubility. Due to sudden oversaturation of uric acid in the urine, uric acid precipitates as crystals or sludge in tubules and collecting ducts. These precipitates cause tubular obstruction and acute renal failure. Interstitial fibrosis or tophus formation is generally not encountered.
Clinical Manifestation of Acute Uric Acid Nephropathy
Acute uric acid nephropathy is typically asymptomatic, although flank pain can occur if there is significant renal pelvic or ureteral obstruction. The diagnosis should be suspected when acute renal failure develops in any of the above clinical settings in association with marked hyperuricemia (plasma uric acid concentration generally above 15 mg/dL). This is in contrast to most other forms of acute renal failure in which the plasma uric acid concentration is less than 12 mg/dL, with the exception of prerenal azotemia where there is an increase in proximal sodium and urate reabsorption. Diagnosis is facilitated by the characteristic clinical syndrome and with a urinary uric acid/urinary creatinine ratio of >1 mg/mg (or >0.66 mM/mM) [29] and by the presence of urate crystals in the urinary sediment.
Treatment of Acute Uric Acid Nephropathy
Prevention is the best therapy for acute uric acid nephropathy. Patients about to receive chemotherapy or radiation for a malignancy with rapid cell turnover should be pretreated with allopurinol plus fluid loading (with saline and possibly mannitol) to maintain a high urine output (over 2.5 L/day) [58, 59]. Historically, treatment consisted of forced alkaline diuresis (to facilitate solubilizing the urate) and large doses of xanthine oxidase inhibitors (typically allopurinol 300–600 mg/day). Recently, recombinant uricase (rasburicase) has become available, which can be administered intravenously and which effectively lowers serum uric acid levels and corrects acute kidney injury more rapidly than allopurinol [60]. Dialysis can also be used to acutely lower the serum uric acid levels. The natural course is one similar to that for acute renal failure of any etiology with a period of oliguria, followed by partial or complete clinical recovery. However, some degree of residual renal injury/damage is common.
Familial Juvenile Hyperuricemic Nephropathy
FJHN is an autosomal dominant disorder associated with juvenile onset of gout and progressive renal disease. Histologically, the kidney usually shows patchy areas of tubular atrophy and fibrosis, focal interstitial infiltration of lymphocytes and histiocytes, and globally or segmentally sclerosed glomeruli. Associated gross thickening and sometimes reduplication of the basement membrane in distal tubules and collecting ducts have been observed [61–63], but urate crystal deposition is rare. Gout may or may not occur in the individual. Urate production is normal in all cases as judged by urinary excretion on a purine-free diet. A decreased fractional urate excretion (FEur) for age and sex precedes any decrease in the glomerular filtration rate in otherwise apparently healthy patients and suggests a primary defect in urate handling with secondary (or late associated) renal damage. Subjects often are normotensive initially, but hypertension is common as the renal disease progresses. Hemodynamic studies have shown that there is severe renal vasoconstriction, with marked depression in renal plasma flow relative to the glomerular filtration rate.
Recently the disease was shown to be due to a mutation in uromodulin, also known as the gene encoding the THP [25]. The TH protein is produced only by the thick ascending limb tubular epithelial cells in the kidney, raising questions of how this mutation could result in hyperuricemia and renal failure. Interestingly, mutations in uromodulin have also been shown to be the cause of type 2 autosomal dominant medullary cystic kidney disease [25]. Indeed, a recent study also suggests that this latter entity is commonly associated with severe hyperuricemia and clinically mimics the phenotype of familial hyperuricemic nephropathy [25]. However, it is clear that FJHN is a syndrome, not a result of defect in a single gene [64].
The pathogenesis of the renal injury remains to be fully elucidated. Interestingly, medullary cystic kidney disease is known to be associated with salt wasting, but it remains unclear if patients with FJHN also have a salt- or water-wasting defect. Preliminary results in FJHN patients, however, suggest that they have a defect in urine concentration, and this correlated inversely with the serum uric acid levels [15]. Furthermore, mice with the uromodulin mutation also show a mild water- and sodium-wasting phenotype and have evidence for upregulation of sodium transporters in their proximal tubules, as well as a relative defect in urinary urate excretion when adjusted for the sodium excretion [65]. This provides the interesting possibility that the hyperuricemia in FJHN is due to increased proximal sodium and urate reabsorption secondary to renal salt loss.
Diagnosis and Treatment of FJHN
Diagnosis is suggested by a positive family history; the early onset of renal insufficiency in the setting of elevated uric acid levels (often >9 mg/dL) disproportionate to age, sex, or degree of renal dysfunction; and by FEur of <5 %. The grossly decreased mean fractional excretion of uric acid found in affected children is even more striking when compared with the normally high FEur of their healthy counterparts (range from 12 % to 30 %). However, the very low FEur in FJHN increases if renal failure progresses, and consequently in early uremia, the FEur may be normal for a while [61] and thus obscure the diagnosis. Confirmation of FJHN can now be obtained through leukocyte DNA analysis for the mutation [62].
Treatment is largely supportive. Aggressive control of high blood pressure is generally thought to be crucial for a successful outcome. The role of allopurinol (and hence a decrease of hyperuricemia) in ameliorating the progression of the renal disease has been stressed by some researchers [63, 66, 67] but is disputed by others [68, 69]. The efficacy of allopurinol clearly relates to the degree of renal damage at the time of treatment initiation, and patient compliance is also important.
Hyperuricemia in Patients with Chronic Kidney Disease
We have discussed the role of hyperuricemia as a cause of kidney disease; however, hyperuricemia may be also seen in patients with established renal diseases, mainly in the setting of reduced renal clearance of uric acid associated with progressive loss of GFR [70]. As discussed earlier, there is some retention of uric acid with renal insufficiency of various causes, but this is usually mild due to compensatory increases in the fractional excretion of uric acid and an increase in enteric excretion in the gut [9, 10]. Indeed, gouty arthritis is uncommon in CKD. This may relate to the fact that uric acid level above 10 mg/dL is unusual in advanced renal failure and that uremia is known to inhibit neutrophil chemotaxis and function [71], and hence the inflammatory response may be partially subdued.
There has been a long-history controversy whether it is beneficial to lower serum uric acid levels in CKD patients and if so, what target level of uric acid needs to be achieved. Asymptomatic hyperuricemia in CKD patients was regarded biologically inert and was not an indication of aggressive hypouricemic therapy without a history of gouty arthritis or biopsy-proven chronic uric acid nephropathy or excessively high serum uric acid level. However, many recent studies provide new evidence that uric acid has contributory role in hypertension and renal and cardiovascular disease, which may change the therapeutic guideline of hyperuricemia in different population of subjects.
There is compelling evidence that hyperuricemia is an independent risk factor for CKD in the general population [46]. In one study, risk for CKD in a subject with hyperuricemia was greater than for that associated with proteinuria [72]. Hyperuricemia has also been reported to be an independent risk factor for renal disease progression in patients with glomerular diseases [73–75] and contrast-induced nephropathy [76] and also may confer increased risk in subjects with essential hypertension [77]. Additional evidence that hyperuricemia may have a role in kidney disease was provided by a prospective clinical trial in 54 hyperuricemia CKD patients with random assignment of allopurinol or placebo for 1 year. Allopurinol treatment resulted in a decrease in serum uric acid associated with a significant slowing of renal disease progression. [48]. Another study reported that diabetic patients with elevated serum uric acid levels were at highest risk for developing progressive nephropathy [78]. In this study, a multiple logistic regression model revealed a significant relationship between uric acid and prevalence of hypertension [odds ratio (OR) = 1.8; 95 % confidence interval (CI) 1.6–2.0] after adjustment for age, sex, and duration of diabetes. The prevalence of macroalbuminuria was significantly higher in patients with hyperuricemia than in those with hypo- and normouricemia. Uric acid was also independently associated with macroalbuminuria, after adjustment for age, sex, HbA1c levels, creatinine clearance, duration of diabetes, and blood pressure levels (OR = 1.5; 95 % CI 1.1–2.0). There have been several recent epidemiologic studies demonstrating the role of uric acid in development of new kidney disease and/or worsening renal function in subjects with different clinical background and comorbidities [79–83].
Besides the effect of hyperuricemia on renal disease progression, an elevated uric acid is also regarded as a cardiovascular risk factor. Given the high prevalence of cardiovascular morbidity in CKD patients, clinical implication of hyperuricemia on cardiovascular complication in CKD subjects may need to be readdressed. A recent prospective study in 294 newly diagnosed patients with CKD stage 5 followed for an average of 6 years revealed that subjects with markedly increased serum uric acid levels (≥9.0 mg/dL) had a twofold increased risk for mortality after adjusting for numerous comorbidities [84]. Although high uric acid levels were associated with lipid levels, calcium/phosphate metabolism, and levels of inflammation markers, an elevated uric acid level itself may represent a true risk factor for cardiovascular disease and mortality in CKD patients [85, 86]. In a randomized, prospective study in 113 patients with estimated GFR (eGFR) <60 ml/min, Goicoechea et al. demonstrated that allopurinol (100 mg/day) was able to slow the progression of renal disease after a mean time of 23.4 ± 7.8 months [87]. No changes in blood pressure or in albuminuria induced by allopurinol have been observed. Interestingly, allopurinol treatment also reduced cardiovascular and hospitalization risk in these subjects.
Potential Mechanisms of Uric Acid-Induced Promotion of Chronic Kidney Disease and Cardiovascular Disease
According to the recent experimental data, uric acid may induce preglomerular arterial disease, renal inflammation, and hypertension via an activation of RAS and COX-2 [51, 88]. There is still limited information, however, about the pathogenic mechanism to verify the causative role of uric acid in renal and cardiovascular diseases. Uric acid is also a mitogen for vascular smooth muscle cells. Rat aortic vascular smooth muscle cells showed de novo expression of COX-2 mRNA after incubation with uric acid [88]. Incubation of the smooth muscle cells with either a COX-2 inhibitor or with a TX-A2 receptor inhibitor prevented a proliferation response to uric acid. COX-2 was also shown to be expressed de novo in the preglomerular vessels of hyperuricemia RK rats, and its expression correlated both with the uric acid levels and with the degree of smooth muscle cell proliferation. These findings suggest a critical role for uric acid-mediated COX-2 generated thromboxane in vascular smooth muscle cell proliferation in animal models of chronic progressive renal disease. In addition to COX-2, it is likely that angiotensin II is also contributing to uric acid-induced vasculopathy. Preglomerular vasculopathy in rats with oxonic acid-induced hyperuricemia can be largely prevented by blocking the RAS, and scientists have also reported that uric acid-mediated vascular smooth muscle cell proliferation can be partially inhibited by blocking the angiotensin II type 1 receptor [51]. Therefore, both angiotensin II and COX-2 are involved in the vascular proliferation and inflammation observed in in vitro and in vivo animal studies.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


