CHAPTER 112 Ulcerative Colitis
Ulcerative colitis (UC) is a chronic idiopathic inflammatory disease of the gastrointestinal tract that affects the large bowel and is a major disorder under the broad group of conditions termed inflammatory bowel diseases (IBDs). Dr. Samuel Wilks is credited with being the first to describe UC in 1859 when he wrote on “idiopathic colitis” and recognized it as distinct from the then more common bacillary dysentery.1 He also reported the pathologic finding of dilated and thinned colon with severe pancolonic inflammation in a patient with this condition.2 In 1909, Hawkins described the chronic and relapsing nature of the disease course and the “stealthy hemorrhage” onset of distal disease, in which bleeding often occurred in the presence of constipation.3 In that same year, Sir Arthur Hurst gave a more complete description of UC, including its sigmoidoscopic appearances and differentiation from bacillary dysentery.4
EPIDEMIOLOGY
In general, there has been a distinct north-south gradient in risk. The areas with the highest rates of reported incidence and prevalence of UC include North America, England, northern Europe, and Australia (Tables 112-1 and 112-2). In North America, incidence rates range from 6.0 to 15.6 cases per 100,000 person-years, and the prevalence ranges from 38 to 246 cases per 100,000 persons.5 In Europe, incidence rates range from 1.5 to 20.3 cases per 100,000 person-years, with prevalence of 21 to 243 cases per 100,000 persons.5 The disease initially had been considered more common in northern Europe, although studies now suggest that the incidence of UC in southern Europe is comparable to that in northern Europe.6 In contrast, studies have reported significantly lower incidence rates of 0.6 to 6 per 100,000 person-years in other parts of the world, including Asia, Africa, and Latin America.5
Table 112-1 Incidence of Ulcerative Colitis in Various Geographic Regions
| REGION | PERIOD OF STUDY | INCIDENCE (PER 100,000 PERSON-YEARS) |
|---|---|---|
| North America | ||
| Alberta | 1981 | 6.0 |
| Manitoba | 1987-1996 | 14.3-15.6 |
| California | 1980-1981 | 10.9 |
| Minnesota | 1984-1993 | 8.3 |
| Europe | ||
| Scandinavia | 1980-1999 | 9.2-20.3 |
| Great Britain | 1985-1994 | 13.9 |
| Northern Europe | 1988-1994 | 3.2-11.8 |
| Southern Europe | 1980-1994 | 1.5-9.6 |
| Asia | ||
| India | 1999-2000 | 6.0 |
| Japan | 1991 | 1.9 |
| Korea | 1992-1994 | 1.2 |
| Africa | ||
| South Africa | 1980-1984 | 0.6-5.0 |
| Central and South America | ||
| All countries | 1987-1993 | 1.2-2.2 |
Adapted from Loftus EV. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004; 126:1504.
Table 112-2 Prevalence of Ulcerative Colitis in Various Geographic Regions
| REGION | PERIOD OF STUDY | PREVALENCE (PER 100,000 PERSONS) |
|---|---|---|
| North America | ||
| Alberta | 1981 | 37.5 |
| Manitoba | 1994 | 169.5 |
| Minnesota | 2001 | 246 |
| Europe | ||
| Scandinavia | 1987 | 161.2 |
| Great Britain | 1995-1996 | 122-243 |
| Northern Europe | 1984 | 24.8 |
| Southern Europe | 1988-1992 | 21.4-121 |
| Asia | ||
| Israel | 1980-1985 | 55.2-70.6 |
| India | 1999 | 44.3 |
| Japan | 1991 | 18.1 |
| Korea | 1997 | 7.6 |
| Singapore | 1985-1996 | 6.0 |
Adapted from Loftus EV. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004; 126:1504.
The overall incidence of UC has remained relatively stable since the 1980s; however, there appears to be geographic variation in this time frame. Although the incidence rates in North America, England, and Sweden have remained unchanged or have declined, those in southern Europe have increased.7 These time trends contrast with those of Crohn’s disease, which has shown an increase in incidence across geographic regions.
There appears to be a marked ethnic variation in the incidence of UC. One ethnic group with high incidence of this disease is the Jewish population. Incidence rates of UC among Jews have been shown to be several-fold higher than in the non-Jewish population across various geographic regions. In the United States, for example, the annual incidence of UC among Jews is 13 cases per 100,000 person-years compared with 3.8 per 100,000 among non-Jewish whites.7 Within Israel, Ashkenazi Jews have a higher incidence than do Sephardic Jews, but they have a lower incidence than in the United States and northern Europe, suggesting that environmental factors also play an etiologic role.
UC traditionally has been considered extremely uncommon in minority populations, but recent studies have challenged this notion. Early studies reported that UC was rare in blacks. Most of these studies, however, were conducted in regions with limited black populations, and more-recent studies suggest an increasing incidence of UC among African Americans. By the late 1970s, incidence rates were comparable between whites and nonwhites in the United States.8,9 An increase in incidence of UC also has been observed among blacks in South Africa, although the incidence rate still remains lower than that for South African whites.10
In Asia, UC is generally is less common than in the Western countries. The prevalence and annual incidence rates for UC in Japan have remained relatively stable at about 5.5 cases per 100,000 persons and 0.36 to 0.5 cases per 100,000 persons, respectively11; this stability is in contrast with the rising incidence of Crohn’s disease in the Japanese population. Limited data also suggest similar findings in the Chinese and Korean populations.12,13 The prevalence of UC in India has been reported to be substantially lower than that among Europeans,14 although an accurate assessment of its epidemiology is hampered by several of the previously mentioned issues. In contrast to the limited data on indigenous South Asians, several studies have demonstrated that South Asian immigrants in England are more likely to have UC than are European natives.14–16 This changing epidemiology with a population emigrating from a low-risk to a high-risk geographic region supports the concept of environmental influences on development of disease.
ETIOLOGY AND PATHOGENESIS
GENETICS
Family History
Genetic factors have been linked to the development of UC, supported largely by the observation that family history is one of the most important risk factors for developing the disease. A familial incidence of UC has been recognized for many years, and although figures vary widely in different studies, about 10% to 20% of patients have at least one other affected family member.17 Familial associations generally occur in first-degree relatives. The relative risk of the same disease in a sibling of a person with UC has been estimated to be between 7% and 17% based on North American and European studies. Parents, offspring, and second-degree relatives appear to be at a lower risk for developing UC than are first-degree relatives. Data from the United States suggest a preponderance of parent-sibling combinations, but in the United Kingdom, the disease is shared more commonly by siblings. Indeed, the strongest evidence of a genetic influence for UC is derived from twin studies. In three large European twin pair studies, approximately 6% to 16% of monozygotic twin pairs had concordant UC compared with 0% to 5% of dizygotic twin pairs.18–20 These concordance rates are substantially lower than those for Crohn’s disease, suggesting that genetic determinants, although important, play a less-significant role for UC than for Crohn’s disease. No twin pair demonstrated both UC and Crohn’s disease, further supporting the genetic basis of these disorders.
Familial association is greater in persons of Jewish descent, a heritage known to have a higher incidence of IBD. The lifetime risk of developing disease is three-fold higher among first-degree relatives of Jewish patients compared with relatives of non-Jewish patients.21 A similar increase in risk also has been observed in relatives of patients who had early onset of disease. This familial association contrasts with the low incidence of UC among spouses of patients with IBD in most series. Although reports of IBD in both spouses are rare, a study of 30 conjugal instances of IBD (17 of which were concordant for Crohn’s disease, 3 for UC, and 10 mixed) found that the majority of these couples developed IBD after cohabitation, thus suggesting a shared environmental exposure.22
For all affected first-degree relatives within a family, there is a high concordance for type of disease (UC vs. Crohn’s disease) and occurrence of extraintestinal manifestations.23 In fact, epidemiologic studies in families with multiple affected members have demonstrated disease-type concordance rates of 75% to 80%, the other 20% to 25% of families being mixed (i.e., having members with both UC and Crohn’s disease).24 The overlap in inheritance patterns among the mixed families suggests that a subset of genes associated with IBD confers susceptibility to both UC and Crohn’s disease, whereas others are specific to one disease or the other.
Numerous studies have examined the effect of family history on disease location. In UC, no consistent correlation has been observed in the studies that examined disease extent namely, left-sided colitis versus extensive colitis and family history of UC.25
Genetic Mutations
The inheritance of UC cannot be described by a simple mendelian genetics model. It is likely that multiple genes are involved and that different genes confer susceptibility, disease specificity, and phenotype. Linkage studies have suggested that there are susceptibility genes for UC on chromosomes 1, 2, 3, 5, 6, 7, 10, 12, and 17.26–29 The IBD2 locus on chromosome 12 appears to have strong linkage demonstrated in studies involving large numbers of families with UC.28 The NOD2/CARD15 gene mutations located on chromosome 16 associated with Crohn’s disease have not been associated with UC, although UC patients from families with a history of Crohn’s disease and UC might possess NOD2 variants.30 In contrast, the C3435T polymorphism for the human multidrug resistance 1 (MDR1) gene is linked to susceptibility for UC but not Crohn’s disease.31 The MDR1 gene product, P-glycoprotein, is highly expressed in intestinal epithelial cells and serves an important barrier function against xenobiotics. In contrast to NOD2/CARD15, the frequency of this polymorphism in patients with Crohn’s disease is similar to that in control subjects.
Genome-wide association studies have looked for overlap of Crohn’s disease genes in patients with UC by using a nonsynonymous single nucleotide polymorphism (SNP) scan.32,33 The Crohn’s disease autophagy genes ATG16L1 on chromosome 2q37 (which encodes autophagy-related 16-like protein) and IRGM on chromosome 5q33 (which encodes immunity-related GTPase [guanosine triphosphatase] family, M), both of which are involved in bacterial processing and the protection of cells from various bacterial pathogens and their toxins (i.e., autophagy), are not seen in patients with UC.32 A number of Crohn’s disease loci or genes (or both) have been identified in UC, however, and include IL-23R on chromosome 1p31, which encodes the interleukin[IL]-23 receptor; chromosome 3p21, which encodes MST1 and other potential genes of interest; IL-12β on chromosome 5q33, which encodes the IL-12 receptor β1 subunit (also known as p40) that constitutes part of both the IL-23 and IL-12 receptors; NKX2-3 on chromosome 10q24, which encodes NK2 transcription factor related, locus 3; and chromosome 17q21, which encodes STAT3 and other potential genes of interest.
Another genome-wide association study has observed a strong association between the gene encoding IL-23R and both Crohn’s disease and UC.33 Several polymorphisms of this gene have been identified, most notably the Arg381Gln polymorphism. Heterozygous carriage of the glutamine allele is associated with a three-fold decreased risk of Crohn’s disease and a more-modest reduction in the risk of UC in non-Jewish populations; this reduction is not seen in UC within the Jewish population. IL-23R is important because it plays a key role in the differentiation of a relatively newly discovered subset of T cells called Th17 cells (see later).
ENVIRONMENTAL FACTORS
Numerous clinical and experimental observations have suggested involvement of intestinal bacterial microbiota in the pathogenesis of IBD. The most obvious observation perhaps is that Crohn’s disease and UC preferentially occur in regions of the bowel that contain the highest concentration of bacteria, namely, the terminal ileum and the colon, where bacterial concentrations approach 1012 organisms per gram of luminal contents. Interestingly, diverting the fecal stream in patients with Crohn’s disease can treat and even prevent disease, whereas reinfusion of ileostomy contents leads to new inflammatory changes within only one week.34 Other human data have shown that antibiotics are useful in the treatment or postoperative prevention of Crohn’s disease and pouchitis. Finally, probiotics (discussed later in this chapter) have been shown to have efficacy in the primary and secondary prevention of pouchitis. The most glaring evidence of the necessary role of bacteria in the pathogenesis of IBD from rodent data is that genetically susceptible mice or rats in a gnotobiotic (germ-free) environment do not have intestinal inflammation; however, these same rodents rapidly develop intestinal inflammation after bacterial colonization.35–38 Just as in humans, rodent gut inflammation can be treated and prevented with antibiotics and probiotics.39,40
With respect to the human gastrointestinal microbiome, much has been learned in the last few years. Complex mathematical models have estimated that the human gastrointestinal microbiome contains at least 1800 genera and between 15,000 and 36,000 species of bacteria41–43; three studies have identified more than 45,000 bacterial small-subunit (SSU) rRNA genes, a technique that captures at most only 50% of the predicted species-level biodiversity.41–43 Of the bacterial genes identified to date, almost all (more than 98%) can be grouped into four phyla: Firmicutes accounts for 64% of the total and includes the family Lachnospiraceae (e.g., Clostridium groups XIVa and IV) and the subgroup Bacillus (e.g., Streptococcaceae and Lactobacillales). Bacteroidetes account for 23% of the total, Proteobacteria account for 8% of the total and include the family Enterobacteriaceae (e.g., Escherichia coli), and Actinobacteria account for 3%.43
Four general mechanisms have been postulated to explain how components of the normal intestinal microbiome might initiate or contribute to the development of the chronic inflammatory state.44 First, microbes can induce intestinal inflammation, either by adhering to or invading intestinal epithelial cells, thereby causing downstream proinflammatory cytokine production or by producing enterotoxins.
Second, a breakdown in the balance between protective and harmful intestinal bacteria, termed dysbiosis, can lead to disease. Most studies comparing the intestinal microbiome in IBD with that in healthy controls show biodiversity in the IBD populations is decreased by 30% to 50%. One study found that this reduction in biodiversity was due to decreased concentrations of Firmicutes (specifically Lachnospiraceae) by 300-fold and Bacteroides by 50-fold.43 The loss of these organisms is important because they are known to produce short-chain fatty acids, such as butyrate, which nourish colonocytes. As a result of their decrease, the relative concentrations of Proteobacteria and Actinobacteria increase in IBD patients relative to controls, although quantitative PCR analysis showed that the absolute numbers of Enterobacteriaceae were not higher in IBD patients than in controls. Loss of protective bacteria, however, could set the stage for overgrowth of pathogenic bacteria. Another interesting finding in this study is that the microbiota of patients with Crohn’s disease and UC were similar to each other. In addition, the study did not identify any individual species that was particularly prevalent in grossly abnormal diseased tissue, and thus no active bacterial etiologic agent was suspected of causing or propagating disease.
In addition to infectious agents, several other environmental factors have been proposed as contributing etiologic factors of UC. The best characterized environmental factor associated with UC is cigarette smoking. Numerous studies have consistently shown that UC is more common among nonsmokers than among current smokers, with the relative risk of UC in nonsmokers ranging from two to six45; this association is independent of genetic background and gender. Furthermore, there may be a dose-response relationship, with the disease more common in current light smokers than in heavy smokers. This relationship is consistent with observations that clinical improvement with nicotine therapy in patients with UC appears to be limited to those treated with high doses of nicotine and not those receiving lower doses. This risk of developing UC with smoking is particularly high for former smokers, especially within the first two years of smoking cessation. The rebound effect also is higher for former heavy smokers than for former light smokers.46 Smokers also appear to have reduced rates of hospitalization for UC and reduced rates of pouchitis following colectomy.47 Studies on the role of passive smoking in UC have yielded conflicting results. A recent meta-analysis of 10 studies examining this issue found no association between passive smoking and future development of UC.48
Several mechanisms have been postulated to account for the apparent protective effect of active smoking on UC. These include modulation of cellular and humoral immunity, changes in cytokine levels, increased generation of free oxygen radicals, and modification of eicosanoid-mediated inflammation. Smoking also might have an effect on mucus production by the colonic mucosa, and might alter colonic mucosal blood flow and intestinal motility. No single mechanism, however, can explain the clinical observation of the beneficial influence of smoking on UC and its adverse effect on Crohn’s disease (see Chapter 111).
Other environmental risk factors that have been suggested to influence the development of UC include diet (wheat, maize, cow’s milk, refined sugar, fruits and vegetables, alcohol), oral contraceptives, food additives (silicon dioxide), toothpaste, and breast-feeding7; none, however, has been shown conclusively to be associated with UC. Studies have suggested an inverse relationship between appendectomy and the subsequent development of UC49,50; the mechanisms for this protective effect of appendectomy on UC are unknown. It is possible that removal of appendiceal-associated lymphoid tissues abrogates certain pathologic alterations in mucosal immune responses and therefore prevents the onset of UC.
IMMUNE FACTORS
Humoral Immunity
Histologic examination of the inflamed colon indicates a marked increase in the number of plasma cells. This increase is not uniform among cells producing different classes of immunoglobulins. The largest proportional increase occurs in immunoglobulin (Ig)G synthesis, which has the highest pathogenic potential among antibody classes. The increase in IgG synthesis in UC is most pronounced in the IgG1 and IgG3 subclasses, in contrast to Crohn’s disease, in which the increase in IgG2 synthesis is more prominent.51,52 This disparity in the local IgG subclass response likely reflects differences in antigenic stimuli or host immunoregulatory responses between the two groups of IBD patients. The increased IgG synthesis in IBD may represent polyclonal stimulation; patients with UC often have circulating antibodies to dietary, bacterial, and self antigens that are mostly of the IgG isotype, usually the IgG1 subclass. Many of these antibodies are thought to be epiphenomena because the serum antibody titers do not correlate with clinical features. Nevertheless, the known cross-reaction between enterobacterial antigens and colonic epithelial epitopes may be an important triggering event, even though, later in the course of the disease, the serum antibody titer to either the bacterial or the colonic antigen may be unimportant.
The concept that UC is an autoimmune disease is supported by its increased association with other autoimmune disorders, including thyroid disease, diabetes mellitus, and pernicious anemia.53 Patients with UC have varying levels of autoantibodies to lymphocytes, ribonucleic acid, smooth muscle, gastric parietal cell, and thyroid; these are specific for neither tissue nor disease. Antibodies to epithelial cell-associated components, which specifically recognize intestinal antigen, also have been described.53 The best-characterized intestinal autoantigen is a 40-kDa epithelial antigen found in normal colonic epithelium.54 This autoantigen is recognized by IgG eluted from the inflamed colonic mucosa of patients with UC and is a component of the tropomyosin family of cytoskeletal proteins.55 The antibody response to this 40-kDa protein appears to be unique to UC and is not found in Crohn’s disease nor in other inflammatory conditions. This autoantigen shares an epitope with antigens found in the skin, bile duct, eyes, and joints, sites often involved in the extraintestinal manifestations of UC. The precise pathogenic significance of this autoantibody in UC, however, remains unclear at present.
An autoantibody that has received significant attention in UC patients is pANCA.56 This autoantibody is present in 60% to 85% of patients with UC.57,58 It is synthesized within the lamina propria and is of the IgG1 subclass. The antigen to which the pANCA is directed has not yet been determined with certainty, and a variety of putative antigens have been proposed, including nuclear histone and nonhistone proteins. The most recent evidence suggests that the antigen is a 50-kd nuclear envelope protein that is specific to myeloid cells.59
Just as with other autoantibodies found in patients with UC, the pathogenic relevance of pANCA in this disorder is unknown. In fact, the prevailing thought is that pANCA has no pathogenic role in UC but that it might serve as a potential marker of susceptibility and genetically distinct subsets of UC. The level of pANCA titer does not correlate with disease activity, but it might decline in patients with long-standing remission or in patients who have had colectomy at least 10 years previously. Studies have suggested that pANCA may be associated with a more-aggressive disease course60 and with the development of pouchitis after ileal pouch-anal anastomosis (IPAA) in patients with UC.61,62 Also of interest is that Crohn’s disease patients with colonic disease have higher rates of pANCA seroreactivity than those without colonic disease, suggesting a possible UC-like Crohn’s disease phenotype.63
Another type of autoantibody commonly seen in IBD, especially in Crohn’s disease, is that against bacterial antigens. Antibodies to bacterial antigens seen in UC include anti-CBir1 and anti-OmpC. Anti-Cbir1 is an antibody to flagellin from Clostridium species; it is found in about 6% of UC patients64 and also appears to be associated with the development of pouchitis.65 Anti-CBir1 also is found in 50% of patients with Crohn’s disease, in which it is associated with more complicated disease. 64 Anti-OmpC (outer membrane porin C of E. coli) is seen more often in UC patients who have a mixed family history of Crohn’s disease and UC rather than those with a family history of only UC.66
Cellular Immunity
Activation of the TLRs and NLRs results in downstream activation of nuclear factor-κB (NF-κB), which then stimulates the transcription of genes coding for various proinflammatory cytokines (including TNF, IL-1, IL-6, and IL-8), chemokines, adhesion molecules, and costimulatory molecules. In addition, activation of NF-κB stimulates the maturation of dendritic cells, which are involved in antigen presentation. Defects in any of the PRR pathways can lead to abnormal bacterial processing and possibly IBD.67
The adaptive immune system, which is governed by T cells and B cells, is specific. Mucosal lymphocytes often are divided into two groups based on location: lamina propria lymphocytes and intraepithelial lymphocytes (IELs). Lamina propria lymphocytes express surface adhesion molecules, α4β7, that provide a homing signal for peripheral immune cells to the mucosal sites.68 Most investigators have found a similar distribution of T-cell subsets (CD4+, CD8+) within the lamina propria in patients with UC compared with that in controls.69 Also, the absolute number of IELs is normal or reduced in UC, most of which are CD8+ cell. The function of IELs has not been well characterized, but it has been suggested that they are cytotoxic and perhaps active in suppressing local immune response. Regardless of their functional status, mucosal T cells within the lamina propria and epithelium, as well as peripheral blood T cells, display a variety of activation markers, suggesting an activated memory phenotype.70 Studies have suggested that the T-cell receptor repertoire is altered in active IBD.71
Although T-cell–mediated immunity has attracted the most attention in the pathogenesis of UC, nonspecific cellular immunity also is altered. In patients with active disease, there is an overproduction of circulating monocytes as well as mucosal macrophages.72 The inflamed mucosa of patients with UC also exhibits infiltration of substantial numbers of granulocytes.
Epithelial Cells
Intestinal epithelial cells serve barrier functions and play a role in enteric immunity. Colonocytes express class II major histocompatibility complex (MHC) antigens and can function as antigen-presenting cells.73 In addition, they also express cytokine receptors, secrete various cytokines and chemokines, and express leukocyte adhesion molecules.74–77 Thus, abnormalities in colonic epithelial cells can contribute to the development of UC.
Patients with UC have an increased turnover rate of colonic epithelium78 and other abnormalities of epithelial cells including reduced metabolism of short-chain fatty acids, especially butyrate, abnormal membrane permeability,79 and altered composition of glycoprotein mucus produced by the colonic epithelium.80 Specifically, the mucus layer in UC appears to be thinner than normal.81 These and other abnormalities can lead to the finding of increased numbers of adherent bacteria, in both the mucus layer and even at the epithelial surface, in patients with UC.82–84 The role of epithelial cells in the pathogenesis of IBD is supported further by animal models of colitis produced by disruption of colonic epithelium.85
Consequences of Immune Activation
The Th1 response is characterized by cell-mediated immunity and is associated with the production of interleukin (IL)-2 and interferon (IFN)-γ. The differentiation of T cells along a Th1 pathway is stimulated by IL-12 generated in response to exposure to infectious agents. The Th2 response is characterized by the production of cytokines IL-4, IL-5, IL-10, and IL-13, which amplify the humoral immune response. Th1 and Th2 subsets reciprocally down-regulate each other through cytokine production.86 Th1 and Th2 pathways can be regulated by unique regulatory T cell (Treg) subsets that produce IL-10 and transforming growth factor (TGF)-β and down-regulate inflammation.87
Historically, the oversimplified view of adaptive immunity in IBD is that Crohn’s disease is mediated by Th1 cells whereas UC is mediated by Th2 cells; there is considerable evidence that the story is much more complex. Macrophages in the inflamed colon of patients with active UC synthesize IL-1β, TNF, and IL-6, whereas lamina propria T cells probably produce IL-2 and IFN-γ. This immune response can be up-regulated further by presentation of antigen to CD4+ lymphocytes by colonic epithelial cells that express HLA class II antigens.73 Studies have implicated a specialized type of T cell, the natural killer (NK) T cell, which seems to mediate the Th2 response in UC.88,89 These NK T cells, which are not classical NK T cells in that they do not express the typical NK T-cell receptors seen with classical NK T cells, secrete large amounts of IL-5 and IL-13 and are actually cytotoxic for intestinal epithelial cells.
A novel T-cell–mediated inflammatory pathway, which appears to be involved in the pathogenesis of both Crohn’s disease and UC, has been discovered and centers on the Th17 cell lineage. Th17 cells have been shown to produce a variety of cytokines, most notably IL-6 and IL-17. IL-17 is a potent proinflammatory cytokine that not only facilitates T-cell activation but also stimulates a variety of cells, including fibroblasts, macrophages, epithelial cells, and endothelial cells, to produce an array of proinflammatory cytokines, including IL-1, IL-6, TNF-α, and chemokines.90 Th17 cell development is inhibited by Th1 and Th2 cells but is promoted by IL-6, TGF-β, IL-21, and IL-23R.91 IL-23R, which is highly expressed by activated Th17 cells, also is expressed by NK cells, NK T cells, other CD4+ T cells, and CD8+ T cells.29 The interaction of IL-23 with its receptor has been shown to have a central role in the development of inflammation in various mouse models of colitis.92,93 Antibodies to IL-23 thus represent another potential therapeutic strategy for the treatment of IBD.
Release of these various cytokines from the T-cell inflammatory pathways also might lead to other abnormalities seen in UC, such as increased epithelial cell permeability and collagen synthesis. Alteration of endothelium by a variety of cytokines can result in local ischemia. Increased expression of endothelial adhesion molecules in response to inflammatory mediators recruits circulating granulocytes and monocytes to the inflamed tissues, thus further perpetuating the inflammatory response. Elevated cytokine levels within the mucosa also stimulate the release of metalloproteinase from fibroblasts with subsequent matrix degradation. Mucosal concentrations of many mediators have been shown to be elevated in patients with active UC, including leukotrienes, thromboxane, platelet-activating factor, nitric oxide, and reactive oxygen metabolites.94 These mediators, which are mostly released from active macrophages and neutrophils, contribute to inflammation and mucosal injury and alter epithelial cell permeability, thereby further contributing to diarrhea. Diarrhea in UC also is caused by complement activation and the release of kinins and other inflammatory mediators from mast cells and eosinophils (see Chapter 2).
PSYCHOGENIC FACTORS
Psychosomatic factors first were implicated in the pathogenesis of UC in the 1930s,95 but there is no good direct evidence to support this concept. Since the introduction of glucocorticoids for the treatment of patients with UC and the focus on immunologic aspects of the pathogenesis of the disease in the 1950s, this previously widely held notion has diminished in popularity.
Experimental studies have helped identify mechanisms of the proinflammatory potential of stress in animal models of colitis.95 When rats are exposed to stress before proinflammatory stimuli are introduced, the severity of colonic inflammation is increased. This particular response has been shown not to be mediated by either vasopressin or corticotropin-releasing factor. In addition, stress has been shown to directly increase intestinal permeability in rats, an action mediated by cholinergic nerves, and to potentiate intestinal inflammation in this particular situation. There are indeed studies reporting that psychosocial stress increases the risk of relapse in patients with quiescent UC.96,97 Conversely, many of the psychological features observed in patients with UC are likely secondary to this chronic disease process, a phenomenon physicians must be aware of when managing these patients.
PATHOLOGY
At the time of initial presentation, approximately 45% of patients with UC have disease limited to the rectosigmoid, 35% have disease extending beyond the sigmoid but not involving the entire colon, and 20% of patients have pancolitis.98 The disease typically is most severe distally and progressively less severe more proximally. In contrast to Crohn’s disease, continuous and symmetrical involvement is the hallmark of UC (Fig. 112-1), with a sharp transition between diseased and uninvolved segments of the colon.
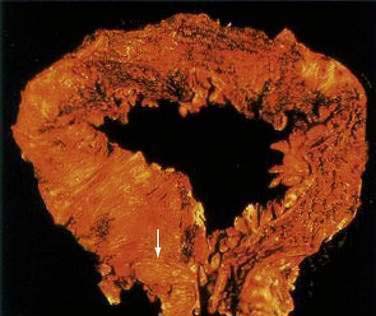
There are a few exceptions to this general rule. First, medical therapy can result in areas of sparing. For example, topical enema therapy can lead to near-complete mucosal healing in the rectum and distal sigmoid colon. Second, up to 75% of patients with left-sided UC have periappendiceal inflammation in the colon and patchy inflammation in the cecum,99 resembling the skip pattern characteristic of Crohn’s disease. These patterns of rectal sparing and skip lesions can lead to a misdiagnosis of Crohn’s disease.
Macroscopically, the mucosa in UC appears hyperemic, edematous, and granular in mild disease. As disease progresses, the mucosa becomes hemorrhagic, with visible punctate ulcers. These ulcers can enlarge and extend into the lamina propria. They often are irregular in shape with overhanging edges or may be linear along the line of the teniae coli. Epithelial regeneration with recurrent attacks results in the formation of pseudopolyps, which is typical of long-standing UC but which also may be seen in acute disease (Fig. 112-2). Another characteristic appearance of long-standing disease is atrophic and featureless colonic mucosa, associated with shortening and narrowing of the colon. Patients with severe disease can develop acute dilatation of the colon, also characterized by thin bowel wall and grossly ulcerated mucosa with only small fragments or islands of mucosa remaining. With perforation of the colon, a fibrinopurulent exudate may be seen on the serosal surface of the bowel.

During the healing phase of UC, the inflammatory infiltrate subsides and epithelial regeneration takes place. Epithelial cells undergoing regenerative changes become cuboidal with eccentric, large nuclei, and prominent nucleoli. These features may be confused with dysplasia. Thus, a diagnosis of dysplasia in UC should be made with caution in the presence of acute inflammation. Accordingly, surveillance colonoscopy (see “Dysplasia and Colorectal Cancer”) should be performed during a period of remission.
A classic histologic feature of chronic quiescent UC is crypt architectural distortion or actual dropout of glands (Fig. 112-3). Architectural changes include branching or bifid glands, wide separation among glands, and shortened glands that do not extend down to the muscularis mucosa. Architectural alteration is a prominent feature of chronic quiescent UC, but the histologic abnormalities can revert to normal after mild flares early in the course of disease. Another characteristic feature of chronic quiescent UC is Paneth cell metaplasia, with Paneth cells located distal to the hepatic flexure, where they normally are absent. Other nonspecific chronic changes seen in UC include neuronal hypertrophy and fibromuscular hyperplasia of the muscularis mucosa. Varying degrees of acute or chronic inflammation of the lamina propria may be present in chronic quiescent disease. A thin band of predominantly lymphocytic inflammation occasionally may be seen deep to the muscularis mucosa, presenting diagnostic challenges.
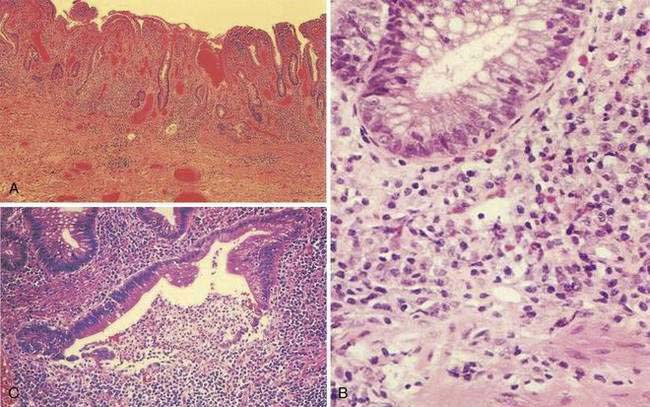
Most of these pathologic findings are not specific for UC. Features that reflect chronicity and thus argue against a diagnosis of infectious or acute self-limited colitis include distorted crypt architecture, crypt atrophy, increased intercrypt spacing to fewer than six crypts per millimeter, an irregular mucosal surface, basal lymphoid aggregates, and a chronic inflammatory infiltrate.100,101 The histologic severity of inflammation does not necessarily correlate with clinical disease activity in patients with UC, because patients may be relatively symptom free although histology reveals significant inflammation.
CLINICAL FEATURES
Patients with UC can present with a variety of symptoms. Common symptoms include diarrhea, rectal bleeding, passage of mucus, tenesmus, urgency, and abdominal pain. In more-severe cases, fever and weight loss may be prominent. The symptom complex tends to differ according to the extent of disease.102 Patients with proctitis often have local symptoms of tenesmus, urgency, mucus, and bleeding, whereas patients with extensive colitis usually have more diarrhea, weight loss, fever, clinically significant blood loss, and abdominal pain. In general, the severity of the symptoms correlates with the severity of the disease; however, active disease may be found at colonoscopy in patients who are otherwise asymptomatic. Additionally, patients with known UC can have severe symptoms that are not necessarily due to UC, such as those caused by bacterial (e.g., Clostridium difficile) or viral (e.g., cytomegalovirus) infections or a host of other similar disorders.
The onset of UC typically is slow and insidious. Symptoms usually have been present for weeks or months by the time the typical patient seeks medical attention. The median interval between the onset of symptoms and diagnosis of UC is approximately nine months.103 Some patients with UC present much more acutely, with symptoms mimicking acute infectious colitis. Indeed, it is not uncommon to find a patient whose UC began after a documented gastrointestinal infection, such as Salmonella or C. difficile. This observation raises the question whether the infection revealed preexisting but silent disease or whether it was actually the initiating factor.
SYMPTOMS
Rectal Bleeding
Rectal bleeding is common in UC, its characteristics determined by the distribution of disease. Patients with proctitis usually complain of passing fresh blood, either separately from the stool or streaked on the surface of a normal or hard stool.104 This symptom often is mistaken for bleeding from hemorrhoids. In contrast to hemorrhoidal bleeding, however, patients with ulcerative proctitis often pass a mixture of blood and mucus and might even be incontinent. Patients with proctitis also often complain of the frequent and urgent need to defecate, only to pass small quantities of blood and mucus without fecal matter. When the disease extends proximal to the rectum, blood usually is mixed with stool or there may be grossly bloody diarrhea. When disease activity is severe, patients typically pass liquid stool containing blood, pus, and fecal matter. This stool often is likened to anchovy sauce, and some patients with this symptom do not actually recognize that they are passing blood. Unless the patient has severe disease, passage of blood clots is unusual and suggests other diagnoses such as a tumor. Active UC that is sufficient to cause diarrhea almost always is associated with macroscopically evident blood. The diagnosis needs to be questioned if visible blood is absent.
Diarrhea
Diarrhea is common but not always present in patients with UC. Up to 30% of patients with proctitis or proctosigmoiditis complain of constipation and hard stools.104 Most patients with active disease complain of frequent passage of loose or liquid stools and may have nocturnal diarrhea. Fecal urgency, a sensation of incomplete fecal evacuation, and fecal incontinence also are common, especially when the rectum is severely inflamed. Diarrhea in this setting often is accompanied by passage of large quantities of mucus, blood, and pus.
The pathophysiology of diarrhea in UC involves several mechanisms, but failure to absorb salt and water is the predominant factor105 and results from reduced Na+,K+-ATPase (adenosine triphosphatase) pump activity, increased mucosal permeability, and altered membrane phospholipids. High mucosal concentrations of lipid inflammatory mediators, which are detected in UC, have been shown to stimulate chloride secretion in normal colon, and it is possible that these mediators also contribute to diarrhea by increasing mucosal permeability. Urgency and tenesmus, which are common symptoms when the rectum is inflamed, are caused by decreased rectal compliance and loss of the reservoir capacity of the inflamed rectum.106 With severe inflammation, the urgency can be sufficiently acute to cause incontinence.
Colonic motility is altered by inflammation, and there is rapid transit through the inflamed colon. With left-sided disease, distal colonic transit is rapid, but there is actual slowing of proximal transit,107 which might help explain the constipation that is commonly seen in patients with distal colitis. Prolonged transit in the small intestine also occurs in the presence of active colonic inflammation.107
LABORATORY FINDINGS
Related posts:
 Surgical Peritonitis and Other Diseases of the Peritoneum, Mesentery, Omentum, and Diaphragm
Surgical Peritonitis and Other Diseases of the Peritoneum, Mesentery, Omentum, and Diaphragm
 Hepatitis Caused by Other Viruses
Hepatitis Caused by Other Viruses
 Hemochromatosis
Hemochromatosis
 Gastrointestinal and Hepatic Complications of Solid Organ and Hematopoietic Cell Transplantation
Gastrointestinal and Hepatic Complications of Solid Organ and Hematopoietic Cell Transplantation
 Nutritional Assessment and Management of the Malnourished Patient
Nutritional Assessment and Management of the Malnourished Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


