CHAPTER 74 Hemochromatosis
Trousseau was the first to describe a case of hemochromatosis in the French pathology literature in 1865.1 Almost 25 years later, in 1889, von Recklinghausen, thinking that the disease was a blood disorder that caused increased skin pigmentation, introduced the term hemochromatosis.1 In 1935, Sheldon published a description of all 311 cases of the disease that had been reported in the world’s literature to that point, including several from his own records. He recognized that hemochromatosis was an inborn error of iron metabolism and that all the pathologic manifestations of the disease were caused by increased iron deposition in the affected organs.1 In 1976, Simon and coworkers2 demonstrated that the gene for hereditary hemochromatosis (HH) was linked to the HLA region on the short arm of chromosome 6. The benefit of early diagnosis on survival was shown in a classic paper by Niederau and colleagues,3 who demonstrated that if HH was identified and treated before the development of cirrhosis or diabetes mellitus, survival of affected patients was equivalent to that of an age- and gender-matched population.
In 1996, the HFE gene was identified on chromosome 6, thereby permitting genetic testing for the two major mutations (C282Y, H63D) that are responsible for HFE-related HH.4 Several prospective population studies have shown that the frequency of the C282Y homozygous state is approximately 1 in 250 in white populations of northern European descent.5 It is now recognized that C282Y homozygosity has incomplete clinical penetrance, with a strong male predominance for symptomatic disease.6 HH is characterized by increased intestinal iron absorption that results from low expression of the iron-regulatory protein hepcidin.5–7 In addition to the discovery of HFE and hepcidin, several additional genes and proteins involved in the regulation of iron homeostasis have been identified, contributing to a better understanding of cellular iron uptake and release. Also, numerous clinical and pathophysiologic studies have been performed and have led to improved diagnosis, better family screening, and new insights into normal and abnormal iron homeostasis. HFE-related HH is a common autosomal recessive disorder of iron metabolism; if it is diagnosed early and treated appropriately, every patient with the disorder can have a normal lifespan.
CAUSES OF IRON OVERLOAD
Hereditary hemochromatosis comprises several inherited disorders of iron homeostasis characterized by increased intestinal iron absorption that results in tissue iron deposition (Table 74-1). The older terms primary hemochromatosis and idiopathic hemochromatosis should no longer be used. The liver is always the principal recipient of most of the absorbed iron and is always involved in symptomatic HH. The most common form of HH by far is HFE-related HH.5–9 It is an autosomal recessive disorder usually identified in adults of northern European ancestry. Most patients who present with HH are homozygous for the C282Y mutation of HFE, although some persons who are compound heterozygotes (C282Y/H63D) also have iron overload.
Table 74-1 Iron Overload Conditions
| Hereditary hemochromatosis | HFE-related hereditary hemochromatosis (type 1) |
| C282Y homozygosity | |
| C282Y/H63D compound heterozygosity | |
| Other HFE mutations | |
| Non–HFE-related hereditary hemochromatosis: | |
| Hemojuvelin (HJV) mutations (type 2A) | |
| Hepcidin (HAMP) mutations (type 2B) | |
| Transferrin receptor 2 (TFR2) mutations (type 3) | |
| Ferroportin (SLC40A1) mutations (type 4) | |
| Loss-of-function mutations | |
| Gain-of-function mutations | |
| African iron overload | |
| Secondary iron overload | Iron-loading anemias |
| Aplastic anemia | |
| Chronic hemolytic anemia | |
| Pyridoxine-responsive anemia | |
| Pyruvate kinase deficiency | |
| Sideroblastic anemia | |
| Thalassemia major | |
| Parenteral iron overload | |
| Iron-dextran injections | |
| Long-term hemodialysis | |
| Red blood cell transfusions | |
| Chronic liver disease | |
| Alcoholic liver disease | |
| Hepatitis B | |
| Hepatitis C | |
| Nonalcoholic steatohepatitis | |
| Porphyria cutanea tarda | |
| Portacaval shunt | |
| Insulin resistance syndrome with iron overload | |
| Dietary iron overload | |
| Miscellaneous | Aceruloplasminemia |
| Congenital alloimmune hepatitis (neonatal hemochromatosis) | |
| Congenital atransferrinemia |
Other inherited forms of iron overload, classified as non-HFE-related HH, are juvenile hemochromatosis and iron overload resulting from mutations in the genes for hepcidin,10 transferrin receptor 2 (TFR2),11 or ferroportin.12 Juvenile HH is characterized by rapid iron accumulation. Mutations in two different genes have been shown to cause forms of juvenile HH. The more common mutation occurs in the HJV gene on chromosome 1q; this gene encodes a protein called hemojuvelin.13 Mutations in the hepcidin gene (HAMP) also produce a form of juvenile HH10; hepcidin is a hepatic peptide that acts to down-regulate iron absorption (see later). Mutations of the gene TFR2 produce an autosomal recessive form of HH that is clinically similar to HFE-related HH.11 How these TFR2 mutations result in iron overload is not yet known; they possibly cause abnormal iron sensing by hepatocytes, the predominant site of TFR2 expression.14 A rare autosomal dominant form of HH results from two categories of mutations in the gene for the iron transporter ferroportin.15 “Loss-of-function” mutations decrease the cell surface localization of ferroportin, thereby reducing its ability to export iron. The result is iron deposition primarily in macrophages, and this disorder is sometimes termed ferroportin disease. The second category includes “gain-of-function” ferroportin mutations that abolish hepcidin-induced ferroportin internalization and degradation; the distribution of excess iron is similar to that in HFE-related HH, primarily parenchymal.
African iron overload occurs primarily in sub-Saharan Africa and is now considered to be the result of a non-HFE–related genetic trait that can be exacerbated by dietary iron loading.16 Some persons who manifest African iron overload consume an iron-rich fermented maize beverage, but iron overload also can occur in people who do not drink this beverage. In most cases, iron-loaded Kupffer cells are prominent in African iron overload; by contrast, Kupffer cells are relatively spared in HFE-related HH. A similar form of iron overload has been suggested to occur in African Americans,17 and further investigations are needed to determine the genetic basis, prevalence, and clinical consequences of this condition.
Persons who absorb excessive amounts of iron as a result of an underlying cause other than any of the previously mentioned inherited defects have secondary iron overload18 (see Table 74-1). Examples are persons with disorders of ineffective erythropoiesis, liver disease (in some cases), increased oral intake of iron, or the rare condition congenital atransferrinemia. Both HH and secondary iron overload should be distinguished from parenteral iron overload, which is always iatrogenic and which leads to iron deposition that is found initially in the reticuloendothelial system. In patients with ineffective erythropoiesis who require red blood cell transfusions, parenchymal and reticuloendothelial iron overload coexists because these people have a stimulus to increased iron absorption and receive iron in the form of red blood cell transfusions. Congenital alloimmune hepatitis is responsible for most cases of neonatal hemochromatosis.19 In these cases, immune-mediated liver injury in the fetus is associated with the development of iron overload. Treatment with intravenous immunoglobulin during pregnancy markedly slows or prevents the development of this condition.
PATHOPHYSIOLOGY
INTESTINAL IRON ABSORPTION
An increase in intestinal iron absorption is a pathogenic characteristic of HFE-related HH.5–9 Understanding the pathogenesis of HH, therefore, requires a review of the determinants of duodenal iron absorption. Because there are no important physiologic mechanisms to regulate iron loss, iron homeostasis depends on a tight linkage between body iron requirements and intestinal iron absorption. Nearly all absorption of dietary iron occurs in the duodenum, where iron may be taken up as either ionic iron or heme.14,20 The absorption of both forms of iron is increased in patients with HFE-related HH.
Absorption of ionic iron across the enterocyte occurs in two stages: uptake across the apical membrane and transfer across the basolateral membrane (Fig. 74-1A). Before uptake, ionic iron must be reduced from the ferric to the ferrous state; this step is accomplished by ferric reductases that are expressed on the luminal surface of duodenal enterocytes. The ferrous iron crosses the apical membrane via divalent metal transporter 1 (DMT-1). Iron taken up by the enterocyte may be stored as ferritin (and excreted in the feces when the senescent enterocyte is sloughed) or transferred across the basolateral membrane to the plasma. This latter process occurs via the transporter, ferroportin. The basolateral transfer of iron requires oxidation of iron to the ferric state by the ferroxidase hephaestin. Uptake of heme occurs by a transporter whose identity remains uncertain. Once internalized, the heme is degraded and the liberated iron is handled by the enterocyte in the same manner as absorbed ionic iron. Patients with HFE-related HH demonstrate increased basolateral transfer of iron from the enterocytes to the plasma. This increased transfer may be the driving force behind the increased intestinal iron absorption that is characteristic of HH. Some studies of patients with HFE-related HH have demonstrated higher duodenal expression of ferroportin and DMT-1.14 The major regulator of intestinal iron absorption is the peptide hormone hepcidin.
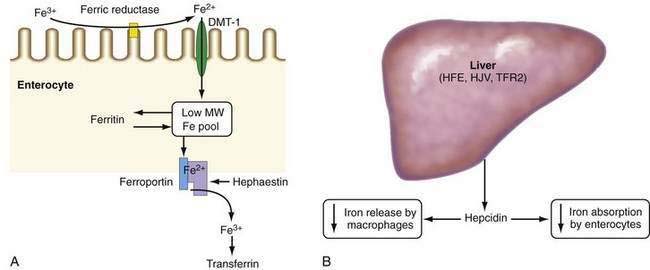
HEPCIDIN
Hepcidin is an iron-regulatory hormone that plays a central role in iron homeostasis by coordinating iron absorption, mobilization, and storage to meet the iron requirements of erythropoiesis and other iron-dependent processes20–23 (see Fig. 74-1B). Hepcidin is expressed predominantly in hepatocytes and is secreted into the circulation. It binds to ferroportin, which is highly expressed on macrophages and the basolateral surface of enterocytes, thereby causing ferroportin to be internalized and degraded, thus inhibiting iron export. Hepcidin expression is regulated by total body iron, erythropoiesis, hypoxia, and inflammation. Excess iron and inflammation induce hepcidin expression, which, in turn, results in decreased intestinal iron absorption and diminished iron release from macrophages. By contrast, hepcidin expression is decreased by iron deficiency, erythropoiesis, and hypoxia, with resulting increases in iron absorption from the intestine and release of iron from macrophages.
In all types of HH, iron overload results from impairment in the hepcidin regulatory pathway. In humans and mice, mutations or knockout of the genes for HFE, hemojuvelin, hepcidin, or TFR2 decrease hepcidin expression, with a resulting increase in intestinal iron absorption via upregulation of ferroportin levels.20–25
Studies have revealed that iron-induced regulation of hepcidin expression involves a bone morphogenetic protein (BMP)-dependent signaling pathway.20,23 BMPs bind to specific receptors on hepatocytes, thereby triggering SMAD protein-dependent activation of hepcidin expression. Selective inhibition of BMP signaling abrogates iron-induced upregulation of hepcidin. Hemojuvelin is a BMP co-receptor and facilitates the binding of BMP to its receptor; knockout of the hemojuvelin gene markedly decreases BMP signaling and hepcidin expression and causes iron overload.
The inflammatory cytokine interleukin-6 upregulates hepcidin via STAT3 (signal transducer and activator of transcription-3) signaling, causing iron retention in macrophages and decreased intestinal iron absorption. The resulting hypoferremia plays a major causal role in the anemia of chronic disease.20–23 Reactive oxygen species inhibit hepcidin expression via a C/EBPα (CCAAT/enhancer binding protein alpha)-mediated mechanism, which may account for the hepatic iron loading associated with alcoholic liver disease and chronic hepatitis C.26
HFE PROTEIN
Studies of HFE protein structure and function were a direct consequence of the cloning of the HFE gene. The HFE gene encodes a 343-amino acid protein consisting of a 22-amino acid signal peptide, large extracellular domain, single transmembrane domain, and short cytoplasmic tail (Fig. 74-2).4 The extracellular domain of HFE protein consists of three loops (α1, α2, and α3), with intramolecular disulfide bonds within the second and third loops. The structure of the HFE protein is similar to that of other major histocompatibility complex (MHC) class I proteins, but evidence indicates that HFE protein does not participate in antigen presentation. Like MHC class I molecules, however, HFE protein is physically associated with β2-microglobulin (see Fig. 74-2). The major mutation responsible for HH results in the substitution of tyrosine for cysteine at amino acid 282 in the α3 loop (C282Y) and abolishes the disulfide bond in this domain.4 Loss of this disulfide bond interferes with the interaction of HFE protein with β2-microglobulin, and the C282Y mutant protein demonstrates decreased presentation at the cell surface, increased retention in the endoplasmic reticulum, and accelerated degradation.27 A second mutation associated with HH results in the change of a histidine to an aspartate at position 63 in the α1 chain (H63D), but this mutation has less biological impact than the C282Y mutation. Like HH patients, HFE-knockout mice manifest higher hepatic iron levels, elevated transferrin saturation (TS), increased intestinal iron absorption, and relative sparing of iron loading in reticuloendothelial cells.14
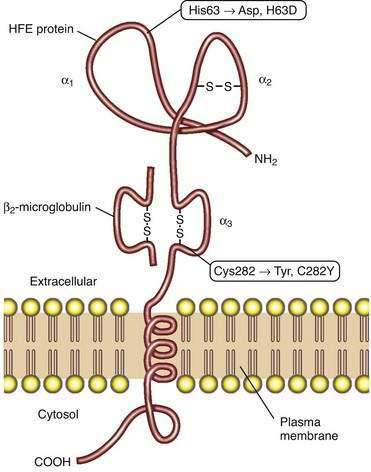
The molecular mechanisms by which HFE influences iron-dependent regulation of hepcidin remain unclear. HFE can bind to both TFR2 and the classic transferrin receptor, TFR1.20 In addition, both HFE and TFR2 may interact with hemojuvelin, suggesting that a complex of HFE and TFR2 may play a regulatory role in BMP signaling. One proposed model suggests that the complex of TFR1 and HFE acts as an iron sensor at the cell membrane of the hepatocyte; as the TS increases, diferric transferrin displaces HFE from TFR1, thereby making HFE available to bind to TFR2. The complex of HFE and TFR2 is then postulated to influence hepcidin expression.20,23









