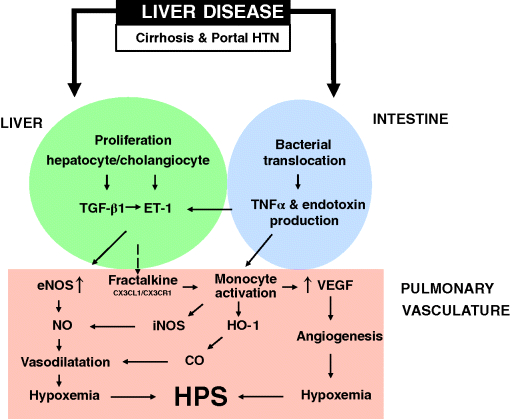
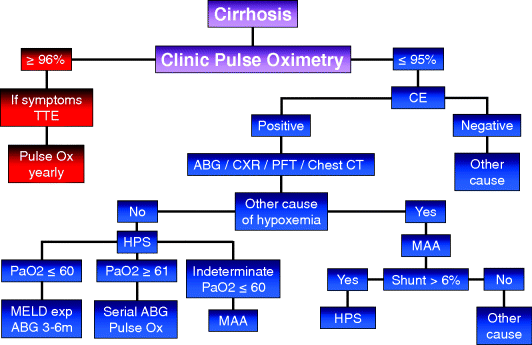
Chapter 17 Moises Ilan Nevah Rubin and Michael B. Fallon Division of Gastroenterology, Hepatology and Nutrition, Department of Internal Medicine, The University of Texas Health Science Center at Houston Houston TX USA Cirrhosis and portal hypertension are associated with alterations in the vasculature in a number of organ systems that alter function in these organs and lead to increased mortality. Although the initial hemodynamic abnormality in cirrhosis is the development of an increased portosystemic gradient (the gradient between the portal vein and the systemic circulation) due to disrupted liver architecture and intrahepatic vasoconstriction, this is promptly followed by splanchnic vasodilatation which, in turn, is followed by systemic vasodilatation. Splanchnic and systemic vasodilatation lead to decreased effective blood volume that activates baroreceptors and neurohumoral systems and is followed by sodium and water retention, increased plasma volume expansion, and cardiac output and, hence, a hyperdynamic circulatory state. Although the development of portal hypertension and a hyperdynamic circulatory state underlie organ dysfunction in liver disease, the specific mechanisms involved appear to vary between organs and are incompletely characterized. One recently recognized end-organ complication of cirrhosis is the hepatopulmonary syndrome (HPS), which results from vascular dilatation and/or angiogenesis in the pulmonary arteriolar circulation [1,2]. The major clinical manifestation is hypoxemia and the presence of HPS has a direct impact on pre- and post-liver transplant mortality [3–5]. The only known cure for the syndrome is liver transplantation. HPS is defined as vascular alterations in pulmonary gas exchange regions, leading to a widened age-corrected alveolar–arterial oxygen gradient (AaPO2) on room air, with or without hypoxemia (AaPO2 ≥15 mmHg, or ≥20 mmHg, in patients ≥65 years old), in patients with underlying liver disease and/or portal hypertension [1]. The diagnosis of HPS requires the presence of a constellation of clinical and laboratory findings and a high index of suspicion. HPS can occur in patients with compensated or decompensated cirrhosis, and has been described in patients with prehepatic portal hypertension (minimal or no evidence of liver dysfunction) and acute liver injury [6–9]. The pulmonary vascular alterations in HPS result from microvascular dilatation and/or angiogenesis. Exclusion of cardiopulmonary diseases has been a key component of the diagnosis in prior definitions. Currently, it is well established that HPS may occur in patients with coexistent cardiac or pulmonary pathologies and be an important contributor to the degree of hypoxemia (Table 17.1) [1,10,11]. Table 17.1 Diagnostic criteria and staging for hepatopulmonary syndrome (HPS). The severity of HPS can be stratified based on the degree of the patient’s hypoxemia as: mild (PaO2 ≥80 mmHg), moderate (PaO2 ≥60 to <80 mmHg), severe (PaO2 ≥50 to <60 mmHg), or very severe (PaO2 <50 mmHg) (Table 17.1). HPS is relatively common in patients with cirrhosis being evaluated for liver transplantation and may occur across the spectrum of severity of liver disease [12–14]. Intrapulmonary vascular dilatation detected by contrast-enhanced echocardiography is present in 40–60% of patients being evaluated for liver transplantation and as many as 30% have sufficient vascular abnormalities to impair gas exchange and cause a widened alveolar–arterial oxygen gradient [5,12,14,15]. Severe HPS, defined as a PaO2 <60 mmHg on room air, is found in 5–15% of patients evaluated for liver transplantation [1]. Although some studies find HPS to be more common in more advanced liver disease and in more severe portal hypertension, it clearly occurs in both compensated cirrhosis and in settings where portal hypertension is present in the absence of cirrhosis. In a prospective multicenter US study, the severity of liver disease was not different between patients with and without HPS [5]. HPS is commonly found in the setting of cirrhosis. It appears to occur less commonly in smokers and may be more common in Caucasians than in Hispanics or African-Americans. HPS has been described in portal hypertension without cirrhosis (prehepatic portal hypertension, nodular regenerative hyperplasia, congenital hepatic fibrosis, and hepatic venous outflow obstruction) [6,7,16] and hepatic dysfunction in the absence of established portal hypertension (acute and chronic hepatitis) [8,9,17]. Intrapulmonary shunting and hypoxemia have also been reported in metastatic carcinoid in the absence of portal hypertension [18] and in children with congenital cardiovascular abnormalities that result in reduced hepatic venous drainage to the lungs [19–21]. Together these observations suggest that factors normally produced or metabolized in the liver may influence the lung microvasculature in susceptible individuals when hepatic function or blood flow are altered. HPS develops when pulmonary microvascular dilatation and/or angiogenesis develops in the setting of liver disease (Figure 17.1). The mechanisms that trigger these microvascular changes are incompletely characterized. In experimental HPS induced by common bile duct ligation (CBDL), both pulmonary microvascular dilatation and angiogenesis contribute to altered gas exchange [2,13]. Similar changes are not observed in other rodent models of cirrhosis and portal hypertension, suggesting that CBDL triggers unique molecular events that influence the pulmonary microvasculature [22–24]. The two most well-recognized events include increased microvascular endothelin signaling and accumulation of monocytes in the microvascular lumen. Upregulated endothelin-1 (ET-1) signaling occurs via increased circulating ET-1 levels, in part derived from cholangiocytes, and by increased lung endothelial endothelin B (ETB) receptor expression in response to increased shear stress due to systemic vascular changes [25–27]. The result is enhanced ETB receptor mediated endothelial-derived nitric oxide (NO) production. Monocyte accumulation within the microvasculature is enhanced by ET-1 alterations and also by tumor necrosis factor α (TNF-α) production from bacterial translocation of intestinal micro-organisms and possibly specific overexpression of monocyte chemotactic and angiogenic chemokines (i.e., fractalkine) [2,23–25,28–31]. Intravascular monocytes are an important source for local pulmonary production of NO, carbon monoxide (CO), and vascular endothelial growth factor (VEGF) which contribute to vascular and gas exchange abnormalities. Recently, in vitro studies have demonstrated that upregulation of the ERK signaling pathway and cholangiocyte VEGF overexpression may be associated with cholangiocyte ET-1 production [32]. Accordingly, in CBDL animals, inhibition of TNF-α, ETB receptor signaling, CO production, tyroxine kinase receptor, or angiogenesis ameliorate HPS [2,23–25,28,29,33]. Figure 17.1 Pathogenesis of the hepatopulmonary syndrome. CO, carbon monoxide; eNOS, endothelial nitric oxide synthase; ET-1, endothelin-1; HO-1, heme oxygenase-1; HTN, hypertension; iNOS, inducible nitric oxide synthase; NO, nitric oxide; TGF-β1, transforming growth factor β1; TNFα, tumor necrosis factor α; VEGF, vascular endothelial growth factor. Less is known about the pathogenesis of human HPS and how the mechanisms identified in the development of experimental HPS contribute to human disease. The fact that HPS occurs across a spectrum of etiologies and disease and portal hypertension severity and develops in less than 50% of cirrhotic patients, suggest that HPS develops in patients with an underlying predisposition. The recent finding that genetic variation in genes associated with vascular growth and development is associated with the risk of HPS raises the possibility that genetic susceptibility to angiogenesis might be a predisposing factor [34]. Systemic and pulmonary production of NO is increased in HPS, but is also increased in cirrhotic patients without HPS [35–38]. Inhibition of NO production does not reliably improve HPS despite improving systemic hemodynamics [35–38]. Therefore, the precise role of NO in human HPS is unknown. CO production does appear to be selectively increased in human HPS, although whether this derives from lung production or influences the vasculature is not known. Finally, whether monocytes adhere in the lung vasculature in human HPS, via increased circulating levels of adhesion molecules, and whether inhibition of TNF-α or bacterial translocation across the gastrointestinal tract alter the severity of HPS are not well defined [28,33,39,40]. The pathogenesis of altered oxygenation in HPS derives from the changes in the pulmonary microvascular bed. In humans, capillary dilatation and pleural arteriovenous malformations have been described in a limited number of cases [6,7,19,20,41]. In experimental HPS, both pulmonary vascular dilatation and angiogenesis occur [2,42]. Three mechanisms of hypoxemia have been described in human HPS: anatomic arteriovenous shunting, ventilation–perfusion mismatch (increased capillary blood flow), and diffusion–perfusion mismatching (impaired passage of oxygen from the alveolus into the vasculature). The relative contribution of these mechanisms to gas exchange abnormalities appear to vary based on the severity of HPS [43]. The majority of patients with HPS are either asymptomatic, particularly if discovered during evaluation for liver transplantation (LT), or develop the insidious onset of dyspnea [44]. Classically, dyspnea (platypnea) and hypoxemia (orthodeoxia) increase in the upright position in HPS due to the predominance of vasodilatation in the lung bases and the increased blood flow through these regions when upright [45]. However, these findings are uncommon and are of limited diagnostic utility [46,47]. Several other clinical features including spider angiomata, digital clubbing, and cyanosis, are also commonly described in HPS but are also not reliable diagnostic indicators. In addition, respiratory symptoms are common in cirrhosis due to poor physical condition, smoking, ascites, and/or intrinsic lung disease. Therefore, the presence of HPS may be difficult to discern and the diagnosis delayed and identified only after severe arterial hypoxemia has ensued. Finally, sleep-time oxygen desaturation also appears to occur commonly in patients with HPS and may worsen hypoxemia at night [48]. Chest radiography (CXR), chest computed tomography (CT), and/or pulmonary function tests (PFT) are often performed to evaluate dyspnea in cirrhosis and during LT evaluation. Commonly, CXR is normal in HPS even when hypoxemia is severe. However, lower lobe interstitial markings resulting from dilated vessels may be present and are often confused with pulmonary fibrosis [12,49]. Dilated vessels as well as parenchymal fibrosis may be visible on high resolution chest CT, but its role in the diagnosis of HPS has not been established. PFTs typically demonstrate well-preserved spirometry and lung volumes. However, the diffusing capacity for carbon monoxide (DLCO) is often reduced and may suggest the diagnosis. However, a decrease in DLCO commonly occurs in cirrhosis in the absence of HPS, limiting diagnostic utility [50,51]. The diagnosis of HPS requires a high degree of clinical suspicion and rests on documentation of the presence of arterial gas exchange abnormalities resulting from intrapulmonary vasodilatation in the appropriate clinical setting. The threshold for pursuing the diagnosis is influenced by the presence of specific signs and symptoms, risk factors for intrinsic cardiopulmonary disease, and whether LT is being considered. In patients with risk factors for intrinsic cardiopulmonary disease (smoking and other cardiovascular risk factors, occupational exposure, liver diseases associated with intrinsic lung disease) such diseases are appropriate initial considerations. In patients with clubbing or dyspnea in the absence of risks for intrinsic cardiopulmonary disease and in those being considered for LT, screening for HPS is appropriate and cost-effective [52]. In the latter group it is particularly important to diagnose and differentiate HPS from portopulmonary hypertension, given that the presence of these disorders may influence treatment and LT candidacy and priority. Gas exchange abnormalities are detected by arterial blood gas measurements and quantified by calculating the alveolar–arterial oxygen gradient (AaPO2 >15–20 mmHg based on age) and assessing for hypoxemia (PaO2 <70 mmHg) [1]. The inclusion of mild gas exchange abnormalities (increased AaPO2, PaO2 <70 mmHg, described as early HPS) appears to be important; based on recent findings, mortality is increased even in this subset of HPS patients relative to non-HPS cirrhotics [53,54]. Obtaining arterial blood gases in the sitting position may enhance the detection of arterial deoxygenation in HPS. Pulse oximetry is an established screening modality for detecting hypoxemia and HPS in patients being evaluated for LT. Using a threshold SpO2 value of <96% provides a sensitivity of 100% and specificity of 88% for detecting HPS patients with a PaO2 of <70 mmHg [55,56]. This technique can target the use of tests for HPS to those with a higher risk of disease. Those patients with a pulse oximetry of <96% should undergo further testing with room arterial blood gases and contrast echocardiography to exclude or confirm the diagnosis of HPS. Based on the utility of pulse oximetry for detecting hypoxemia in a wide range of disorders, this technique may also be useful for HPS screening in the general cirrhotic population (Figure 17.2).
The Hepatopulmonary Syndrome
Introduction
Definition
Cirrhosis and/or portal hypertension
Age corrected alveolar–arterial oxygenation gradient ≥15 mmHg or PaO2 <70 mmHg
Intrapulmonary vasodilatation documented on contrast enhanced echocardiography
Absence of abnormalities that can explain hypoxemia (abnormal chest X-ray, pulmonary function tests or CT)
Mild HPS
Alveolar–arterial oxygenation gradient ≥15 mmHg and PaO2 ≥80 mmHg
Moderate HPS
Alveolar-arterial oxygenation gradient ≥15 mmHg and PaO2 <80 – ≥60 mmHg
Severe HPS
Alveolar-arterial oxygenation gradient ≥15 mmHg and PaO2 <60 – ≥50 mmHg
Very severe HPS
Alveolar–arterial oxygenation gradient ≥15 mmHg and PaO2 <50 mmHg
Adapted from the European Respiratory Society task force on PHD diagnostic criteria for HPS, Rodriguez-Roisin et al, 2004 [1]. CT, computed tomography; PaO2, partial pressure of oxygen in the arterial blood.
Epidemiology
Pathogenesis
Clinical Features
Diagnosis
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel







Full access? Get Clinical Tree


