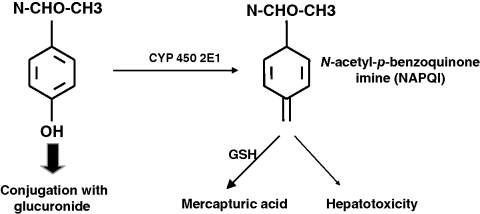
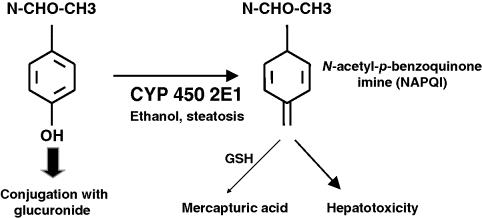
Chapter 23 Felix Stickel Department of Gastroenterology and Hepatology, University Hospital Zürich, Switzerland Administering drugs to patients with acute or chronic liver diseases – either for the underlying liver disease, or to treat nonhepatic conditions – is everyday routine in medical practice. Concerns of physicians that many of the functions that the liver harbors to handle foreign substances could be impaired due to the underlying liver disease are understandable bearing in mind that the liver has a central role in the absorption, distribution, and elimination kinetics of most drugs and many active or inactive drug metabolites [1]. Moreover, the liver is not only the primary site of biotransformation of xenobiotics, but also affects drug distribution, bioavailability, and biliary excretion. In addition, patients with hepatic dysfunction may also be more sensitive to the effects, both desired and adverse, of several drugs, and tolerate an event of drug-induced liver injury (DILI) less well than individuals without pre-existing liver diseases. However, only few methods are available for the assessment of the metabolic function of the liver, and among those that are established, all capture only a proportion of the liver’s function [2]. Avoiding known hepatotoxins and adjustment of dosage of drugs without intrinsic hepatotoxicity in patients with liver dysfunction may therefore be required to avoid excessive accumulation of the drug, active drug metabolite(s) thereof, and subsequent adverse effects, either hepatic or extrahepatic. As no surrogate parameter is currently available to reliably reflect the patency of hepatic metabolism of drugs, their pharmacokinetic characteristics and interactions with other drugs must be considered to avoid therapeutic misadventures in patients with impaired liver function. While this is true for certain substances that are primarily metabolized by the liver, a common misconception prevails that subjects with pre-existing liver diseases are more susceptible to develop DILI than those without. In fact, clinical data supporting this general attitude of caution are limited to only a few drugs, while the majority of prescription and over-the-counter drugs can be used safely. Any foreign compound that enters the human body undergoes metabolism and elimination via exhalation, urinary clearance, or biliary/fecal excretion. Anatomically and functionally, the liver is located between the gastrointestinal tract and the systemic circulation. Drugs (and multiple other molecules contained in the gut) are absorbed predominantly in the small bowel and transported to the liver via the portal vein. Together, the intestinal epithelium and the liver are responsible for the presystemic elimination, the so-called first-pass metabolism of drugs and other xenobiotics before these reach the post-hepatic circulation. Drug-metabolizing enzyme systems are present in many tissues of the body, including the gut epithelia, but their magnitude is expressed in liver tissue. Here, biotransformation takes place by the action of multiple drug-metabolizing enzymes including microsomal cytochrome P450 isoenzymes, mixed-function mono-oxygenases, glutathione-S-transferases, and UDP-glucuronosyltransferases, to name but a few. Some of these can be induced through variable mechanisms which may lead to large interindividual variability in pharmacokinetics and susceptibility for drug-related liver damage [2]. Obviously, the capacity to handle drugs can be hampered with a reduced compartment of metabolically active liver cells such as in severe acute or chronic liver injury. Accounting for this impairment of liver function, pharmacotherapy in clinical hepatology requires the assessment of liver function beyond the determination of routine laboratory biomarkers such as liver-related serum transaminases (alanine aminotransferase, ALT; aspartate aminotransferase, AST) or markers of synthetic (coagulation factors, albumin) or excretory (conjugated bilirubin) function. However, available methods reflecting the metabolic function of the liver are neither satisfactory nor routinely used in clinical practice [3]. Thus, decision making in clinical practice to date relies on data from clinical population-based studies, case reports, spontaneous reporting of adverse events to pharmacovigilance authorities, safety data of registrations trials, and the few randomized trials in which the incidence of DILI in specific cohorts was deliberately sought. In addition to scientific data, information on safety, applicability, and contraindications of certain drugs are provided in the manufacturers’ package inserts of drugs to guide prescribers and users. However, the most important factor in this context is that acting physicians are aware of the true limitations and risks of several drugs in the context of hepatic insufficiency, keep in mind the possibility of DILI even with drugs generally considered safe, and are able to translate this knowledge into clinical judgment [4]. In the following sections, individual drugs associated with a risk of causing DILI are described with a special focus on the context of prescribing them to patients with coexisting liver diseases. Among these, the use of only a few is associated with an increased risk of DILI when administered to patients with pre-existing liver diseases (Table 23.1). Table 23.1 Drugs to avoid or to be used with special precautions in patients with liver diseases. Paracetamol is among the most widely used antipyretic analgesics in the world, and a common cause of fulminant hepatic failure [5], but differs from most other drugs resulting in DILI in that its hepatotoxicity is dose related and thus predictable once the threshold of toxicity has been exceeded. Hepatotoxicity of paracetamol is due to biotransformation of the drug to a toxic metabolite that requires conjugation with glutathione via activity of glutathione transferase to be inactivated [6]. Once glutathione is depleted, the toxic metabolite, N-acetyl-p-benzoquinone imine (NAPQI), binds to other nucleophilic groups in the cell. Cell death ensues, especially in zone III hepatocytes around the terminal hepatic vein, but also in nonliver cells that are able to activate the parent compound such as renal tubular cells, which can lead also to kidney failure. NAPQI formation is enhanced with induction of cytochrome-P450 2E1 (CYP2E1), an isoenzyme responsible for the oxidative degradation of ethanol and other chemicals (Figure 23.1). CYP2E1 can be induced, and thus capable of increased NAPQI production, by excessive alcohol consumption, obesity, and fasting, thereby exhausting glutathione stores required for NAPQI detoxification [7]. Figure 23.1 Under normal therapeutic circumstances, the magnitude of a therapeutic dose (>90%) of paracetamol is glucuronidated (or sulfated) and then excreted. A small percentage is metabolized by cytochrome P450 enzymes (CYP) to the reactive intermediate N-acetyl-p-benzoquinone imine (NAPQI), which is readily detoxified by conjugation with glutathione (GSH) (A). In situations of overdose and/or induced CYP enzymes, as in chronic drinkers or the heavily obese, detoxification mechanisms are exhausted and excessive formation of NAPQI occurs. In addition, glutathione stores may be depleted prior to paracetamol ingestion (malnutrition) aggravating the heaptic insufficiency to detoxify NAPQI (B). Awareness of paracetamol as a potential cause of liver injury is relatively high, but this often leads to its avoidance in patients with liver disease. However, contrary to common belief, paracetamol can be safely used even in patients with cirrhosis, although a maximum safe dose has not yet been established. Data from a randomized trial indicate an increased incidence of liver enzyme elevations up to 20-fold the upper limit of normal in healthy subjects taking 4 g/day paracetamol [8], and an earlier study in regular and excessive alcohol drinkers showed massive elevations of transaminases and fulminant liver failure in 20% of subjects who ingested paracetamol at doses of 4–6 g/day [9]. However, another more recent study in which subjects included consumed one or two alcoholic beverages per day detected only mild elevations of liver enzymes within the normal range following the intake of up to 4 g/day paracetamol [10]. In line with the latter study, Temple et al. [11] demonstrated that patients with impaired liver function neither had higher NAPQI levels nor liver enzyme elevations following repeated doses of 4 g/day paracetamol. Other studies support the view that short-term use of paracetamol at 4 g/day in alcoholics, particularly when abstinent, and subjects with hepatitis C virus (HCV) infection is safe [12,13]. From these data it can be concluded – leaving a margin of safety – that paracetamol dosed at or below 2 g/day can be given to patients with pre-existing liver diseases, whereas doses up to 4 g/day should not be given long term, particularly when regular alcohol consumption is an issue. Nonsteroidal anti-inflammatory drugs (NSAIDs) are potent agents used for the treatment of chronic mild to moderate pain. NSAIDs are among the most widely used drugs worldwide, either via prescription or as over-the-counter preparations. However, non-selective NSAIDs, also known as cyclooxygenase (COX) inhibitors, carry an increased risk of serious upper gastrointestinal complications, including gastroduodenal ulcers, perforation and bleeding, and liver injury. Regarding the latter, along with anti-infectious agents, they top the list for causes of DILI [14]. The incidence of liver disease induced by NSAIDs reported in clinical studies is fairly uniform, ranging 0.3–9 per 100 000 depending on the drug under scrutiny, but considerable under-reporting of asymptomatic, mild cases prevent a precise assessment of the actual risk of DILI along with NSAIDs [15]. Following numerous cases of severe liver injury including fulminant hepatic failure, several NSAIDs, such as bromfenac, ibufenac, and benoxaprofen, have either been withdrawn from the market, or, as for nimesulide, are licensed with tight restrictions [16]. Although liver-related hepatotoxicity induced by NSAIDs generally reveals a low incidence in the general population, several agents appear to carry a higher risk such as sulindac, diclofenac, and nimesulide. While the incidence of NSAID-associated DILI in patients with pre-exsting liver disease has not been systematically investigated, several agents should not be used under certain circumstances: Methotrexate is an efficacious and cost-effective treatment for rheumatoid arthritis and psoriasis, but associated with significant safety issues, particularly regarding hepatotoxicity. Among the risk factors predisposing to methotrexate-related liver injury are female gender, steatosis, regular alcohol consumption, a higher duration and cumulative methotrexatedose, and possibly, cotherapy with acitretin, a synthetic retinoid [23]. However, weighing the risks against the possible benefits of methotrexate, a clear contraindication for its use in patients with liver diseases appears inappropriate, also because noninvasive biomarkers and novel follow-up tools such as transient elastography (Fibroscan™) are available [24]. Bearing this in mind, regular liver biopsies after each 1.5 g of cumulative methotrexatedose – as recommended by several guidelines – seems inordinate, even in those with coexisting liver pathologies. Mok et al. [25] retrospectively investigated the safety of the use of disease modifying antirheumatic drugs (sulfasalazine, chloroquine, gold, methotrexate) in patients with rheumatoid arthritis, either with chronic HBVand HCV infection (n = 30) or without (n = 62), and found that all of the disease-modifier drugs were associated with significantly higher rates of liver enzyme elevations, with a single case HBV reactivation in a patient on azathioprine. In clinical practice, liver enzymes should be monitored during the start of therapy, and HBV carriers be treated with an antiviral drug to prevent reactivation. Particular prudence must be applied when treating patients with novel “biologicals” such as infliximab, etanercept, adalimumab, or certolizumab who are infected with HBV, because reactivation of viral replication and subsequent severe courses of acute hepatitis are well described. In a recent systematic review of published cases, HBV reactivation was reported in 39% of HBsAg+ carriers, and overall more likely to occur in patients previously treated with immunosuppressive agents, and lower in those who received antiviral prophylaxis (23% vs. 62%; P = 0.003). Notably, acute liver failure was reported in five patients, four of whom died [26]. This remarkable risk of biologicals in causing potentially fatal sequelae of acute hepatitis in HBV carriers does not apply to patients with chronic HCV in whom the safety profile of anti-TNF-α agents seems to be acceptable with only minor and rare incidents of liver enzyme elevations [27]. Buprenorphine, a synthetic molecule derived from thebaine, has been marketed in numerous countries as an opiate replacement therapy in drug addicts, a subpopulation of patients often afflicted with chronic infection with HCV and human immunodeficiency viruses (HIV). At recommended sublingual doses, buprenorphine is well tolerated, although a mild increase in serum ALT activity can occasionally occur in some patients. Some heroin addicts inject buprenorphine intravenously and may develop plasma peak concentrations several-fold higher than with sublingual administration. Several case series have reported on acute cytolytic hepatitis with marked jaundice following both intravenous [28] and sublingual administration of buprenorphine [29,30]. Of note, most patients in these series were anti-HCV positive. An interesting report described two HCV-positive individuals who developed acute hepatitis following intravenous buprenorphine, and cleared HCV-RNA after hepatitis subsided [31]. However, more recent and larger series of intravenous drug users receiving opiate replacement therapy with buprenorphine, either infected with HCV or HIV, documented no evidence of hepatotoxicity when buprenorphine was taken in recommended doses [32,33]. Thus, for clinical guidance, patients taking buprenorphine should be advised to take it only via the recommended route, and at doses considered safe. Specific follow-up guidelines are not warranted, but clinicians should take buprenorphine hepatotoxicity into account once liver enzyme elevations occur in a patient. Naltrexone, a long-acting opioid antagonist that possesses no opioid agonist activity, was approved by the US Food and Drug Administration (FDA) for the treatment of opiate dependence in 1984 and alcoholism in 1994. A black box warning by the FDA indicates that naltrexone can cause liver damage and demands that users should be informed about this prior to prescription. However, this view has been challenged by many, and observational studies have not confirmed the hepatotoxicity risk ascribed to naltrexone neither when used in alcoholics nor in patients with HCV coinfection [34,35]. As opposed to naltrexone and buprenorphine, no reports on hepatotoxicity from methadone have been published so far. Derelict alcoholics with advanced alcoholic liver injury or HIV-positive individuals with coexistent HBV or HCV infection may be afflicted by tuberculosis in a substantial proportion, and must be treated with antituberculosis agents. It is important to note that up to 20% of patients receiving isoniazide develop asymptomatic elevation of liver enzymes, often without a tendency to further deteriorate clinically. However, an increased risk for isoniazide hepatotoxicity exists for elderly patients, those who regularly drink alcohol, in malnourished individuals, and in combinations of isoniazide with other antituberculosis drugs [36]. In addition, as described in a previous study by Ungo et al. [37], HIV positive individuals revealed a fourfold higher risk of DILI from antituberculosis drugs, a fivefold higher risk among subjects with chronic HCV, and a remarkable 14.4-fold risk in those coinfected with HIV and HCV. Another study showed similar hazard ratios for patients with chronic HBV infection receiving antituberculosis agents [38].
Agents and Drugs: Precautions in Patients with Cirrhosis
Introduction
Impaired Liver Function and Drug Biotransformation
Drug name/group
Liver disease
Remarks/precautions
Paracetamol/acetaminophen >2 g/day
Alcoholic liver disease/cirrhosis
Can be safely used up to 4 g/day in noncirrhotic patients
“Biologicals” (infliximab, etanercept, adalimumab, certolizumab)
Untreated hepatitis B virus infection
Co-treatment with antiviral agent required (e.g., lamivudine, tenofovir)
“Disease modifiers” (sulfasalazine, chloroquine, gold, methotrexate, azathioprine)
Untreated hepatitis B virus infection
Co-treatment with antiviral agent required (e.g., lamivudine, tenofovir)
Add folate to methotrexate
Isoniazide-containing antituberculosis treatment regimens
Alcoholic liver disease, chronic viral hepatitis B/C, particularly when coinfected with HIV
Regular liver enzyme level monitoring, switch to isoniazide – sparing regimens according to guidelines [37]
HAART
HIV–HCV coinfected subjects
Increased incidence of DILI related to nearly all classes of HAART agents; regular laboratory testing including lactate (lactic acidosis)
Antidepressants
All causes of advanced liver diseases
Avoid MAO inhibitors, tricyclic antidepressants, and nefazodone
Hormonal agents
Tamoxifen
Antiandrogens (flutamide, cyproterone acetate)
Nonalcoholic fatty liver disease/insulin resistance
Chronic viral hepatitis
Regular liver laboratory testing; restriction of use to clear indications
Herbal preparations
All liver diseases
Lack of efficacy and safety data; restrict use to clinical trials
DILI, drug-induced liver injury; HAART, highly active antiretroviral therapy; HCV, hepatitis C virus; MAO, monoamine oxidase inhibitor.
Acetaminophen/Paracetamol
Nonsteroidal Anti-Inflammatory Drugs
Antirheumatic Agents
Opiate Replacement Therapy
Antituberculosis Agents
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


