Androgen receptor (AR)-mediated signaling is critical to the growth and survival of prostate cancer. Although medical castration and antiandrogen therapy can decrease AR activity and lower PSA, castration resistance eventually develops. Recent work exploring the molecular structure and evolution of AR in response to hormonal therapies has revealed novel mechanisms of progression of castration-resistant prostate cancer and yielded new targets for drug development. This review focuses on understanding the mechanisms of persistent AR signaling in the castrate environment, and highlights new therapies either currently available or in clinical trials, including androgen synthesis inhibitors and novel direct AR inhibitors.
- •
Androgen receptor (AR)-mediated signaling is critical to the growth and survival of prostate cancer.
- •
Recent work exploring the molecular structure and evolution of the AR in response to hormonal therapies has revealed novel mechanisms of progression and yielded new targets for drug development.
- •
The vast majority of castration-resistant tumors still rely on AR activity.
Introduction
Prostate cancer is the most common cancer in men and is unique in that its growth is largely dependent on androgen signaling. This dependence was first recognized more than 70 years ago when Huggins and Hodges observed striking regressions of metastatic prostate cancer in men who underwent either surgical or medical castration. Indeed, in men with metastatic prostate cancer, androgen deprivation improves bone pain, lessens lower urinary tract symptoms, and increases overall quality of life.
Despite the impressive clinical improvements observed with androgen deprivation therapy (ADT), androgen suppression is not curative, and all men eventually develop disease growth despite castrate levels of testosterone. Tumor growth in the castrate environment is lethal and accounts for close to 30,000 deaths annually in the United States. The median survival for a man with castrate-resistant disease is between 2 and 3 years from the time of diagnosis of castration resistance.
Until recently, the terms androgen-independent prostate cancer (AIPC) or hormone refractory prostate cancer (HRPC) were used in the literature to describe this clinical state, implying largely that tumor growth occurred through pathways completely independent of the androgen signaling. Work in recent years, however, has dispelled this misconception and has clearly demonstrated that signaling through the androgen receptor (AR) remains crucial to tumor growth in the castrate tumor environment. With this understanding, the term castration-resistant prostate cancer (CRPC) has gradually emerged to describe this clinical state. The term “CRPC” is especially useful in that it connotes only the clinical state of disease growth despite castrate levels of serum testosterone, while not indicating the potential, one way or another, for response to further androgen manipulation.
As we shall see in this review, castration resistance can develop in many ways, including through the development of AR mutations, through AR amplification, through the production of androgens within tumors (intracrine androgen synthesis), through the emergence of truncated AR proteins (splice variants) capable of ligand-independent signaling, and through other mechanisms. Understanding these mechanisms has led the way for the development of multiple novel agents that target the AR itself. Indeed, enzalutamide, a novel antiandrogen that directly inhibits AR function, and abiraterone acetate (Zytiga), an inhibitor of androgen synthesis that lowers circulating ligand, have both been shown to improve overall survival in well-powered randomized Phase III studies and are discussed later in this article. Additionally, a number of new agents are in clinical development, including agents that directly target the AR, such as ARN-509 and EPI-001, as well as those, like abiraterone acetate, that indirectly target the AR, such as orteronel (TAK-700), galeterone (TOK-001), and others.
To understand the clinical development of these agents, we first review our current understanding of the structure and function of the AR, then explore how castration resistance emerges, focusing specifically on mechanisms that lead to persistent AR signaling in a low testosterone environment. We then review the clinical development of agents that target the AR, focusing on recently approved agents and those currently in clinical development.
AR structure and function
The AR gene is located on chromosome Xq11-12 and is a member of the steroid hormone receptor family that includes, among others, the estrogen receptor, progesterone receptor, and the glucocorticoid receptor. All share a similar structure. Histologically, the AR is present in benign prostate epithelial cells as well as in all stages and grades of primary and metastatic prostate cancer. Functionally, the AR is a 110-kDa ligand-activated transcription factor consisting of 917 amino acids that contain 3 important distinct domains ( Fig. 1 A): a carboxy-terminal ligand-binding domain (LBD), which binds androgens, a DNA-binding domain (DBD), and a regulatory N-terminal domain (NTD). Within the N-terminal domain lies the activation function 1 (AF-1) and AF-5 domains, which contain binding sites for transcriptional coregulators and are essential for AR activity.
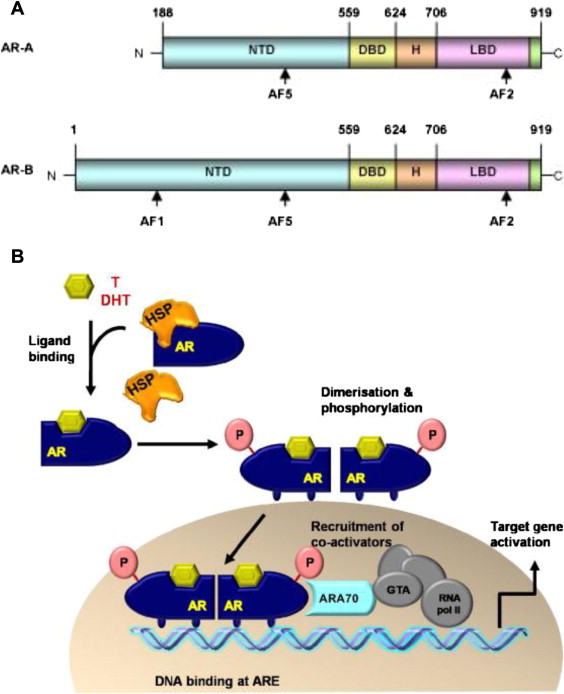
Under normal conditions, inactive AR is found in the cytoplasm of prostate cancer cells and is stabilized there by various heat-shock proteins (HSPs), which expose the LBD to surrounding proteins and allow for androgen binding. Once bound to androgenic ligands, a conformational change occurs in the AR, causing dissociation of HSPs, AR receptor dimerization, and AR migration to the nucleus (see Fig. 1 B). Once inside the nucleus, the AR DBD binds to specific androgen response elements (AREs) on the promoter or enhancer regions of androgen-regulated genes. Transcription of genes necessary for prostate cancer growth and survival can then be initiated, and is enhanced by the binding of transcription coregulators to the AF-1 binding site in the AR-NTD. Among the many genes under AR transcriptional control is prostate-specific antigen (PSA), and hence serum PSA measurement serves as a clinically useful biomarker for AR transcriptional activity and, by extension, disease growth.
AR structure and function
The AR gene is located on chromosome Xq11-12 and is a member of the steroid hormone receptor family that includes, among others, the estrogen receptor, progesterone receptor, and the glucocorticoid receptor. All share a similar structure. Histologically, the AR is present in benign prostate epithelial cells as well as in all stages and grades of primary and metastatic prostate cancer. Functionally, the AR is a 110-kDa ligand-activated transcription factor consisting of 917 amino acids that contain 3 important distinct domains ( Fig. 1 A): a carboxy-terminal ligand-binding domain (LBD), which binds androgens, a DNA-binding domain (DBD), and a regulatory N-terminal domain (NTD). Within the N-terminal domain lies the activation function 1 (AF-1) and AF-5 domains, which contain binding sites for transcriptional coregulators and are essential for AR activity.
Under normal conditions, inactive AR is found in the cytoplasm of prostate cancer cells and is stabilized there by various heat-shock proteins (HSPs), which expose the LBD to surrounding proteins and allow for androgen binding. Once bound to androgenic ligands, a conformational change occurs in the AR, causing dissociation of HSPs, AR receptor dimerization, and AR migration to the nucleus (see Fig. 1 B). Once inside the nucleus, the AR DBD binds to specific androgen response elements (AREs) on the promoter or enhancer regions of androgen-regulated genes. Transcription of genes necessary for prostate cancer growth and survival can then be initiated, and is enhanced by the binding of transcription coregulators to the AF-1 binding site in the AR-NTD. Among the many genes under AR transcriptional control is prostate-specific antigen (PSA), and hence serum PSA measurement serves as a clinically useful biomarker for AR transcriptional activity and, by extension, disease growth.
Evolution of AR structure and function in prostate cancer
Depletion of circulating testicular androgens through either orchiectomy or luteinizing hormone–releasing hormone (LHRH) therapy drastically decreases androgenic ligand levels, reduces AR signaling, lowers PSA, and results in regression of disease the vast majority of cases. Both the rate of PSA decline and the nadir PSA on therapy are predictive of overall response to initial ADT, with higher nadir PSA values and shorter times to PSA nadir associated with poor survival. This observation further highlights the role that AR-mediated signaling has on disease progression and disease aggressiveness.
Antiandrogens, such as bicalutamide (Casodex), nilutamide (Nilandron), and flutamide (Eulexin), have been in clinical use for decades, and observations about their use illustrate one of the first clinically relevant, identifiable steps in AR evolution. It was first noted in the early 1990s that disease progression, despite the combination of an LHRH agonist and an antiandrogen, could be stopped and reversed simply through discontinuation of the antiandrogen. This antiandrogen withdrawal effect (AAWD) was best characterized in a study by Small and colleagues who observed that discontinuation of an antiandrogen in men with a rising PSA leads to sustained PSA declines of more than 75% in more than 10% of men. Further work demonstrated that mutations in the AR can cause AR inhibitors, such as bicalutamide that bind to the LBD, to paradoxically stimulate AR and result in disease growth. Although this mechanism remains incompletely understood, this observation illustrates the point that genomic changes occur or arise in the AR in response to therapy and can have important functional and clinical implications. Whether these genomic changes are present in a subpopulation of cells at baseline, or whether they are acquired in response to the selective pressure of AR-targeted therapy, is still unknown.
Further observation about the state of the AR as tumors transition from castration-sensitive to castration-resistant have had important function implications and have helped guide the development of new therapies. In a landmark study, Stanbrough and colleagues profiled the expression signatures of primary and metastatic prostate tumors, including both castration-sensitive and castration-resistant tumors, and found significantly higher levels of AR mRNA in castrate-resistant tumors compared with castration-sensitive tumors. Subsequent studies of metastatic CRPC tumors obtained at autopsy showed that the genomic segment of chromosome X encoding the AR is itself amplified, up to 60-fold in some cases, and other studies has shown that the AR protein is overexpressed in metastatic CRPC tumors. These observations suggest that depletion of circulating androgens results in a selection for cells that are best able to respond to the low levels of residual ligand.
A second critical laboratory observation is that mRNA encoding enzymes that synthesize hormones are upregulated within tumor cells themselves. This novel finding suggested that tumor cells that were deprived of androgens through the use of an LHRH agonist or through receptor inhibition by an antiandrogen could, in addition to increasing AR number, potentially synthesize their own androgens within the local tumor environment and thereby stimulate their own growth. This finding of autocrine stimulation has been lent further support by the finding of increased levels of androgens within CRPC tumors, even despite low levels of circulating androgen.
Coupling these observations of AR gain and intratumoral androgen synthesis has drastically changed the current thinking about the development of CRPC. In older models, CRPC results from the development of mechanisms of growth independent of the androgen axis. The current model of CRPC is one in which tumors are in effect hypersensitive to even low levels of androgens, and one in which tumors are no longer necessarily reliant on ligand produced by outside sources, such as the testis or adrenal glands. These observations, therefore, provide a plausible mechanism by which formerly castration-sensitive tumors regain the ability to grow in a castrate environment, and provide targets (ie, inhibition of the androgen synthesis machinery) for drug development. Agents either available or in late-stage clinical trials that specifically target androgen synthesis include abiraterone acetate, orteronel (TAK-700), galeterone (TOK-001), and ketoconazole (Nizoral).
Yet hypersensitivity to androgens and upregulated intracrine androgen synthesis is not the whole story. Although AR amplification can be seen in up to 70% of tumors, there are still CRPC tumors that retain a wild-type AR copy number. Similarly, not all CRPC tumors overproduce androgens at a local level. Thus, recent work has explored other mechanisms of autonomous AR activation in men with castrate levels of serum testosterone. Perhaps the most intriguing of these observations is that aberrations can occur in the post-transcriptional splicing of AR mRNA, leading to various forms of a truncated AR protein. Although many forms of these splice-variants are maladaptive and lead to a nonfunctioning AR protein, multiple studies have found the presence, in CRPC tumors, of AR proteins that no longer contains an LBD, but retains the DBD and NTD. Lacking a binding site for ligand, these AR splice-variant mutants can thereby be rendered insensitive to manipulation of circulating or local hormone levels. Despite their insensitivity to ligand, by retaining functional DBDs and NTDs these splice variants nonetheless are able to translocate autonomously to the cell nucleus and cause transcription of AR regulated genes, resulting in truly ligand-independent cell growth and survival.
Cooperation of other signal transduction pathways with the AR, also known as AR transactivation, has also been shown to enhance AR signaling even in the absence of ligand. Specifically, the SRC kinase is a ubiquitous nonreceptor tyrosine kinase involved tumor cell proliferation, survival, and migration, and has been shown to both be upregulated in CRPC cell lines and to cooperate with the AR to enhance AR signaling. Importantly, the SRC kinase can be inhibited with available agents, and in preclinical models inhibition of SRC appears to decrease AR transactivation and downstream AR signaling. Other proteins, such as the cAMP dependent protein kinase (PKA), interleukin-6, and epidermal growth factor, have been implicated in AR transactivation in the absence of ligand through interaction with the AR-NTD. To what degree these pathways enhance AR signaling in the absence of ligand in patients, and whether it is possible to identify the emergence of these pathways in patients in real time to target this pathway, is controversial and the subject of current research. Nonetheless, this work points to another druggable nonandrogen-mediated mechanism of AR activation.
The understanding that the AR can lose sensitivity to circulating androgens through the development of AR splice variants or through AR transactivation has guided the development of novel AR inhibitors with mechanisms that go beyond impairing ligand-receptor interactions, but rather focus on preventing the AR from reaching target sequences in the cell nucleus. Enzalutamide (MDV-3100), ARN-509, and EPI-100 are examples of this new class of and are discussed in further detail later in this article.
To best understand the new agents that have been recently approved or are in clinical development, it is best to think about these agents as a class. This review, therefore, first explores agents that “indirectly” target the AR by inhibiting androgenic ligand production, then focuses on agents that “directly” inhibit interactions between ligand and the AR LBD, and last focuses on agents that impair the interaction of the AR with target sequences in the DNA.
Indirectly targeting the AR: inhibitors of androgen synthesis
Androgen Deprivation Therapy
ADT can be accomplished either through bilateral orchiectomy or medically through the use of gonadotropin-releasing hormone (GnRH) therapy. The GnRH agonists leuprolide (Lupron), goserelin (Zoladex), and triptorelin (Trelstar) are synthetic GnRH analogs that are more than 100 times more potent than natural GnRH and less susceptible to enzymatic degradation. Binding of these agents to receptors in the pituitary gland causes a burst of LHRH release and an initial rise in testosterone production by testicular Leydig cells. Persistent GnRH stimulation over time, however, leads to a downregulation in pituitary GnRH receptors, leading to declines in LHRH secretion and compensatory falls in testicular testosterone production to castrate levels.
Because GnRH agonists cause an initial serum testosterone flare, novel GnRH antagonists, which avoid raising serum testosterone levels, have been developed. Degarelix (Firmagon) was approved by the Food and Drug Administration (FDA) in 2008 and works by inhibiting the interaction of endogenous GnRH with receptors on pituitary gonadotropin-producing cells. Although useful for patients with newly diagnosed widespread metastatic disease in whom a testosterone flare should be avoided, the higher incidence of local injection site reactions (40% vs <1% for men receiving leuprolide) and the need for monthly injections make long-term treatment with this agent less desirable for most practictioners.
Regardless of the method, ADT to achieve a serum testosterone level lower than 50 ng/dL is the mainstay of the treatment of metastatic prostate cancer, and is beneficial when combined with radiation therapy for patients with high-risk localized disease. Recent work has shown that the degree of AR suppression achieved by initial ADT, as measured by serum PSA production, is significantly correlated with the duration of response to hormone therapy. In a study of men receiving ADT for metastatic disease, a time to nadir PSA of less than 6 months was associated with shorter overall survival on univariate analysis. Similarly, the median survival for men with a PSA nadir of 0.2 ng/mL or less was 75 months, compared with 44 months for men with a nadir PSA between 0.2 to 4.0 ng/mL, and only 13 months for men who never nadired below 4.0 ng/mL. These findings support the notion that incomplete suppression of AR signaling leads to AR-mediated disease growth and faster disease progression.
Ketoconazole
Ketoconazole is a synthetic oral imidazole antifungal designed to disrupt fungal cell membranes through inhibition of ergosterol synthesis. Because of the homology between specific fungal and human enzymes, ketoconazole also impairs androgen synthesis in humans, specifically though inhibition of CYP51A, CYP11A1, CYP11B1, CYP11B2, CYP17, and CYP19. Because it also suppresses mineralocorticoid and glucocorticoid synthesis, ketoconazole is given with a replacement dose of corticosteroid, usually oral hydrocortisone. In the largest randomized study to date, ketoconazole was given to men with CRPC at the time of AAWD. PSA responses were observed in 32% of patients taking ketoconazole compared with 10% of men undergoing AAWD alone ( P <.001), with twice as many patients having objective responses in the ketoconazole arm. Although crossover of patients randomized to AAWD alone to ketoconazole likely obscured any overall survival benefit, 2 important observations to come out of this study were that patients who had a more than 50% PSA decline had a 41-month survival, compared with 13 months in those who did not ( P <.001), and that patients with high baseline circulating androstenedione levels were more likely to benefit from therapy than those with low circulating levels. Both of these observations suggest that androgens remain important even after the development of castration-resistance, and show that decreasing the levels of other androgenic ligands in tumors still reliant on ligand-receptor stimulation can greatly slow the growth of the CRPC. Whether baseline circulating androgen levels can be used to identify a population of patients with CRPC more likely to benefit from ketoconazole is still the subject of debate.
Abiraterone Acetate
The recognition that inhibition of androgen synthesis by ketoconazole could result in both PSA and sustained objective responses led the way for the search for better inhibitors of androgen synthesis. Abiraterone acetate is the prodrug of abiraterone, a potent and selective inhibitor of CYP17 (17alpha-hydroxylase/C17,20-lyase), an enzyme that catalyzes key steps in the synthetic pathway of androgens ( Figs. 2 and 3 ). Abiraterone acetate plus prednisone was tested against placebo plus prednisone in a randomized Phase III study of 1195 men with metastatic CRPC who had previously received docetaxel. After a median follow-up of 12.8 months, a statistically significant survival benefit was observed in the abiraterone/prednisone arm with median survival of 14.8 months in this arm, compared with 10.9 months in the placebo/prednisone arm (hazard ratio, 0.65; P <.001). This study was the first ever to show an overall survival benefit to a “secondary” hormonal therapy, as well as the first ever to show a survival benefit for patients with docetaxel-treated CRPC. With the results of this study, abiraterone acetate received FDA approval in 2011 and has become a standard of care for men with docetaxel-treated CRPC.
Related posts:
 Moving Toward Personalized Medicine in Castration-Resistant Prostate Cancer
Moving Toward Personalized Medicine in Castration-Resistant Prostate Cancer
 Palliative Care in Castrate-Resistant Prostate Cancer
Quality of Life with Advanced Metastatic Prostate Cancer
The Experience with Cytotoxic Chemotherapy in Metastatic Castration-Resistant Prostate Cancer
Palliative Care in Castrate-Resistant Prostate Cancer
Quality of Life with Advanced Metastatic Prostate Cancer
The Experience with Cytotoxic Chemotherapy in Metastatic Castration-Resistant Prostate Cancer
 Management of Docetaxel Failures in Metastatic Castrate-Resistant Prostate Cancer
Management of Docetaxel Failures in Metastatic Castrate-Resistant Prostate Cancer
 Targeting Angiogenesis as a Promising Modality for the Treatment of Prostate Cancer
Targeting Angiogenesis as a Promising Modality for the Treatment of Prostate Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


