Antiangiogenic therapy has been successful for the treatment of solid tumors. Several strategies have been used to target angiogenesis in prostate cancer. These strategies include blocking proangiogenic factors via monoclonal antibodies or small molecule inhibitors targeting downstream signaling effector pathways, or using agents with immune-modulatory effects. This review examines the general concepts of tumor angiogenesis and the key clinical trials that have used these agents and other novel biologics in prostate cancer. Targeting angiogenesis is still a promising treatment strategy in prostate cancer with a rational trial design and combination approach.
- •
Vascular endothelial growth factors (VEGF) and their receptors are key players for angiogenesis in prostate cancer.
- •
Promising early phase clinical trials using a VEGF-specific antibody for the treatment of prostate cancer did not translate an overall survival benefit in a recently reported phase III study.
- •
Despite increased toxicity, targeting angiogenesis in combination with systemic therapy is logical for a future trial design.
- •
Results from a few ongoing phase III studies are eagerly awaited.
- •
Better patient selection and a biomarker-driven study will likely lead the success of antiangiogenic therapy in prostate cancer.
Introduction: angiogenesis and cancer
Angiogenesis, or the process of new blood vessel formation, is necessary during cancer progression. Tumor angiogenesis is recognized as one of the hallmarks of malignancy and is regarded as a prerequisite for local tumor growth and expansion. Although tumor cells may live in an avascular state at a microscopic-sized population either as in situ tumors or as a microcylinder of tumor cells enveloping a preexisting microvessel, further growth or expansion of tumor mass critically depends on recruitment of new vessels, or angiogenic switch. The classical angiogenic switch and features of tumor endothelium and vessel structure is summarized in Box 1 . Induction of the angiogenic switch depends on angiogenic balance, which is orchestrated by a variety of activators and inhibitors (see Box 1 ). The proangiogenic gene expression is increased by physiologic stimuli, such as hypoxia, which results from increased tissue mass, and is also determined by tumor cells. The main components involved in this angiogenic switch are (1) tumor cells that are genetically unstable and manifested by oncogene activation or tumor-suppressor mutation/inactivation, (2) proangiogenesis factors released from tumor cells, and (3) endothelial cells (ECs) that are genetically stable and regulated by proangiogenesis factors. Therapies involving targeting one of these 3 components are of tremendous interests to control cancer and improve survival.
- •
The angiogenic switch is a discrete step in tumor development that can occur at different stages in the tumor progression depending on the nature of the tumor and its microenvironment.
- ○
It has to occur to ensure exponential tumor growth.
- ○
It continues as long as the tumor grows.
- ○
It begins with perivascular detachment and vessel dilation, followed by angiogenic sprouting, new vessel formation and maturation, and the recruitment of perivascular cells.
- ○
It is comprised of cancer cells, proangiogenesis factors, endothelial cells (endothelial progenitor cells in bone marrow or peripheral blood), and pericytes (proangiogenic monocytes and pericyte precursors).
- ○
It is off when the effect of proangiogenic molecules (eg, vascular endothelial growth factors, fibroblast growth factors, platelet-derived growth factor β (PDGFB), epidermal growth factors, lysophosphatidic acid (LPA), and so forth) is balanced by antiangiogenic molecules (thrombospondin-1, angiostatin, endostatin, canstatin, tumstatin, and so forth) and is on when the net balance is tipped in favor of angiogenesis.
- ○
- •
Tumor vascular endothelium is phenotypically different from that of normal vessels and it is characterized by increased fenestration and leakiness, aberrant architecture with arteriovenous shunts, multiple loops, and fan and spiral motifs. It has high vascular permeability without functional lymphatics. It has nonuniform surface markers with delayed maturation.
Vascular endothelial growth factors and their receptor signaling pathways
Proangiogenesis factors activate endothelial-cell proliferation and migration. They are mainly tumor-derived receptor tyrosine kinase ligands, such as vascular endothelial growth factor (VEGF), fibroblast growth factors (FGFs), platelet-derived growth factors (PDGF), and epidermal growth factors (EGF), but can be of other origins.
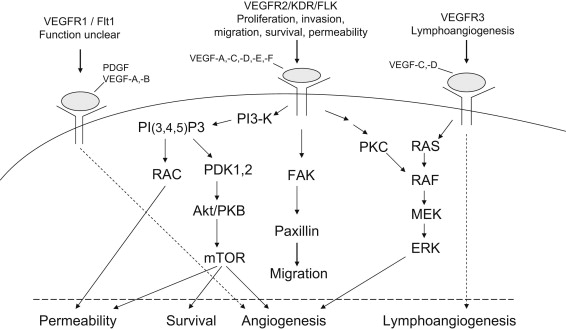
VEGF (also called VEGF-A), originally known as vascular permeability factor, is one of the most potent and well-characterized proangiogenic proteins. Fig. 1 illustrates the family of VEGF molecules and receptors (adapted from Alfiky and Kelly, also see review ). VEGF receptor 2 (VEGFR-2) is the major VEGF signaling receptor that mediates sprouting angiogenesis (called kinase-insert domain–containing receptor [KDR] in humans and fetal liver kinase 1 [flk-1] in mice). The role of VEGFR-1 in sprouting angiogenesis is much less clear. The binding of VEGF to VEGFR-2 leads to a cascade of different signaling pathways (examples of which are shown in the Fig. 1 .), resulting in the upregulation of genes involved in mediating the proliferation and migration of ECs and promoting their survival and vascular permeability. For example, the binding of VEGF to VEGFR-2 leads to dimerization of the receptor, followed by intracellular activation of the phospholipase Cγ (PLCγ)–protein kinase C(PKC)–Raf kinase-MAP/ERK kinase (MEK) mitogen-activated protein kinase (MAPK) pathway and subsequent initiation of DNA synthesis and cell growth, whereas the activation of the phosphatidylinositol 3′–kinase (PI3K)–serine/threonine protein kinase B (Akt) pathway leads to increased endothelial-cell survival. The activation of Src oncogene can lead to actin cytoskeleton changes and the induction of cell migration. The binding of VEGF-C to VEGFR-3 mediates lymphangiogenesis. The understanding of principal molecular pathways for angiogenesis and the role of angiogenesis in cancer progression has resulted in robust research investigation in antiangiogenesis therapy for cancer.
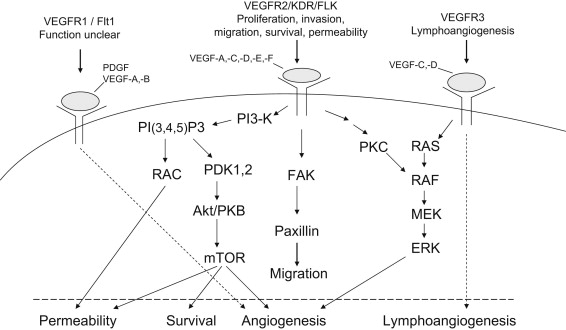
Vascular endothelial growth factors and their receptor signaling pathways
Proangiogenesis factors activate endothelial-cell proliferation and migration. They are mainly tumor-derived receptor tyrosine kinase ligands, such as vascular endothelial growth factor (VEGF), fibroblast growth factors (FGFs), platelet-derived growth factors (PDGF), and epidermal growth factors (EGF), but can be of other origins.
VEGF (also called VEGF-A), originally known as vascular permeability factor, is one of the most potent and well-characterized proangiogenic proteins. Fig. 1 illustrates the family of VEGF molecules and receptors (adapted from Alfiky and Kelly, also see review ). VEGF receptor 2 (VEGFR-2) is the major VEGF signaling receptor that mediates sprouting angiogenesis (called kinase-insert domain–containing receptor [KDR] in humans and fetal liver kinase 1 [flk-1] in mice). The role of VEGFR-1 in sprouting angiogenesis is much less clear. The binding of VEGF to VEGFR-2 leads to a cascade of different signaling pathways (examples of which are shown in the Fig. 1 .), resulting in the upregulation of genes involved in mediating the proliferation and migration of ECs and promoting their survival and vascular permeability. For example, the binding of VEGF to VEGFR-2 leads to dimerization of the receptor, followed by intracellular activation of the phospholipase Cγ (PLCγ)–protein kinase C(PKC)–Raf kinase-MAP/ERK kinase (MEK) mitogen-activated protein kinase (MAPK) pathway and subsequent initiation of DNA synthesis and cell growth, whereas the activation of the phosphatidylinositol 3′–kinase (PI3K)–serine/threonine protein kinase B (Akt) pathway leads to increased endothelial-cell survival. The activation of Src oncogene can lead to actin cytoskeleton changes and the induction of cell migration. The binding of VEGF-C to VEGFR-3 mediates lymphangiogenesis. The understanding of principal molecular pathways for angiogenesis and the role of angiogenesis in cancer progression has resulted in robust research investigation in antiangiogenesis therapy for cancer.
Antiangiogenic therapy versus cytotoxic chemotherapy
The scientific basis of antiangiogenic therapy for cancer rests on 2 general principles: (1) tumor growth is angiogenesis dependent and (2) antiangiogenic therapy targets the genetically stable microvascular ECs. This therapeutic approach was originally thought to be superior to cytotoxic chemotherapy because drug resistance would be less of an issue. Table 1 summarizes the difference between antiangiogenesis therapy and cytotoxic chemotherapy. Antiangiogenesis is the mainstay treatment of recurrent or metastatic renal cell carcinoma and is able to improve the efficacy of chemotherapy for metastatic colorectal cancer, lung cancer, and glioblastoma multiforme (reviewed in ). To date, no antiangiogenic agents have been approved for use in prostate cancer (PCa), although extensive studies on angiogenesis in PCa have revealed that angiogenesis plays a role in the progression of PCa.
| Target | Angiogenesis Inhibitors | Chemotherapeutic Agents |
| Dividing ECs (genetically stable) | Cancer cells (genetically unstable), possible dividing ECs in spouting vessels; endothelial progenitor cells in bone marrow or peripheral blood | |
| Onset | Delayed | Immediate |
| Therapeutic index | Greater, so less toxic | Limited because of side effects (ie, myelosuppression, and so forth) |
| Effect | Cytostatic → stabilization of disease | Cytotoxic or cytostatic → regression of disease or stabilization |
| Tumor regression | Usually no | Yes |
| Maintenance therapy | Likely needed | Usually not needed but proved to be beneficial in some cases |
| Resistance development | Yes | Yes |
Rational of targeting angiogenesis for PCa: role of Hypoxia-induced factor-1α and VEGF in PCa carcinogenesis and progression
By directly measuring P o 2 from the pathologically involved side of the prostate, as well as from a region of normal stromal muscle for comparison, Movsas and colleagues found that hypoxic regions existed in primary human PCa and increasing levels of hypoxia were associated with higher clinical stages. Hypoxia-induced factor-1α (HIF-1α), a key player for angiogenesis, was found overexpressed in many common human cancers and their metastases, including PCa. The significance of HIF-1α in PCa pathogenesis, crosstalk between HIF-1α and androgen receptor (AR) was reviewed by Lin and colleagues. Microvessel density has been used as a surrogate histologic measure of angiogenesis within a tumor. It has been shown to correlate strongly with metastasis potential, Gleason score, and a negative clinical prognosis in PCa.
Fig. 2 illustrates the interplay among hypoxia, HIF-1α, growth factor signaling/autocrine loop, and AR in PCa. Collectively, HIF-1α has emerged as an important transcription factor in PCa biology and is essential in the early stages of prostate carcinogenesis as well as in the transition from the castrate-sensitive (androgen dependent) into the castrate-resistant (androgen independent) stage. Therapeutic actions of antiandrogens or androgen deprivation therapy (ADT) in PCa actually include the inhibition of HIF-1 function. Intratumoral upregulation of HIF-1 contributes to castration resistance by sensitizing AR to a very low concentration of ligand available in the environment. Thus, the inhibition of HIF-1 pathways, which is achieved by many antiangiogenic agents, is able to target AR signaling and provide a therapeutic benefit in the management of PCa.
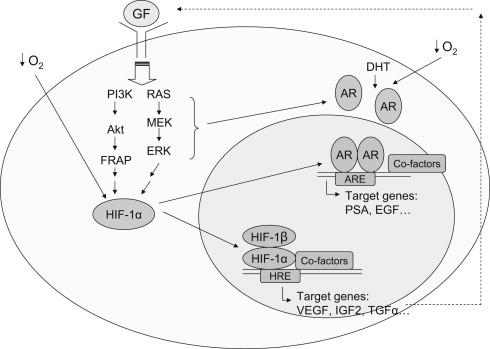
VEGF is expressed by a variety of human solid tumors, including PCa. VEGF plays a critical role in the pathogenesis and progression of human PCa. In human PCa cells, the expression of VEGF receptors (Flk-1/KDR) correlates with poorly differentiated tumors and poor prognosis. VEGF is present in both localized and metastatic prostate tumors, and increasing the plasma concentration of VEGF correlates with metastatic disease progression. In patients with metastatic castration-resistant PCa cancer (mCRPC), both plasma and urine VEGF levels are independent predictors of overall survival (OS). These data supported the role of VEGF in PCa progression and the hypothesis that inhibition of VEGF may enhance current therapies in advanced PCa.
Antiangiogenic agents in clinical development for treatment of PCa
Angiogenesis inhibitors in clinical trials include (1) agents that directly inhibit ECs, (2) agents that block activators of angiogenesis, and (3) agents that block extracellular matrix breakdown. The extracellular matrix of tumors is a complex network of heparan sulfate proteoglycans (HSPGs), collagen, laminin, and fibronectin. Heparanase is one of the antiangiogenesis targets because it is a ubiquitous endoglycosidase that specifically cleaves HSPGs facilitating angiogenesis through growth factor release and metastasis by enhancing cell invasion, migration, intravasation, and extravasation. In PCa, many agents were studied in the preclinical and clinical setting with the hope that either single antiangiogenic agent or in combination with other agents can suppress PCa growth and help patients live longer. Table 2 lists the most known agents in clinical development for PCa.
| Mechanisms and Drugs | Cellular Targets/Mechanisms | Clinical Phase of Development (Reference) |
|---|---|---|
| Anti-VEGF agents Bevacizumab Aflibercept (VEGF-trap) | Humanized IgG monoclonal antibody to all the isoforms of VEGF Fusion protein of the human extracellular domains of VEGFR-1 and VEGFR-2 and the Fc portion of human IgG; binds to both VEGF-A and PlGF with a higher affinity than monoclonal antibodies | III, with docetaxel ± bevacizumab, mCRPC, completed, reported III, docetaxel ± aflibercept, mCRPC, completed, (NCT00519285) |
| Small molecular tyrosine kinase inhibitors Sunitinib (SU11248) Sorafenib (BAY 43–9006) Cabozantinib AZD2171/cediranib | Inhibits the VEGFR, PDGFR, Flt-3, C-kit, CSF-1R, and RET receptor tyrosine kinases Raf kinase (B-Raf, C-Raf) inhibits VEGFR (VEGFR-2 and VEGFR-3), PDGFR, and C-kit MET, VEGFR2, C-RET, and C-KIT VEGFR2&3, FGFR, PDGFR | III, mCRPC with prednisone II, mCRPC, with docetaxel II, mCRPC, after docetaxel II, mCRPC single agent III, mCRPC (NCT01522443) II, NCT00436956 |
| Antiangiogenesis and immunomodulatory agents Thalidomide Lenalidomide | Not fully understood Modulation of integrins and immunomodulatory and antiinflammatory effects, tumor microenvironment | II, mCRPC III, mCRPC, on hold (NCT00988208) |
| Others Tasquinimod Enzastaurin 17-allylamino-17-demethoxygeldanamycin EMD 121 974 (cilengitide, NSC 707 544) PI-88 | Upregulates the expression of thrombospondin-1, downregulates HIF-1α and VEGF Serine/threonine kinase inhibitor of the beta isoform of protein kinase C Inhibitors of HSP90 and promotes degradation of HIF-1α α ν β 3 and α ν β 5 integrin receptor antagonist; inhibits ECs proliferation, migration, adhesion, and ultimately survival Extracellular matrix, inhibitor of angiogenesis and heparanase activity | III, mCRPC (NCT01234311) II, mCRPC II, mCRPC (NCT00118092) II, nonmetastatic mCRPC I/II, mCRPC |
Rational of targeting angiogenesis in combination with systemic therapy
Cancer is driven by an extremely complicated network of signaling proteins. A mutual stimulation occurs between the stroma (ie, angiogenesis) and tumor parenchyma that sustains tumor growth, invasion, and metastasis. Therefore, the tumor cell and vascular system should be considered a functional unit regarding tumor growth. Targeting both tumor cells and angiogenesis is likely more effective than just targeting one of the tumor entities because antiangiogenic and cytotoxic agents work independently on different cellular targets. Preclinical evidence suggests that combining an antiangiogenic agent with a conventional cytotoxic agent or radiation therapy may have an additive or even synergistic antitumor effect. Although the exact mechanism of the additive or synergistic effect is not known, antiangiogenic therapy has been shown to reduce interstitial fluid pressure (IFP). In tumors, there is increased IFP, which forms a barrier to transcapillary transport and is an obstacle for the delivery of therapeutic agents to the tumor site. When IFP is reduced by antiangiogenic therapy, the tissue diffusion of chemotherapy is improved. In addition, conventional cytotoxic chemotherapy drugs can inhibit tumor cell proliferation or may inhibit the growth of several normal host cell types that contribute to angiogenesis pathway. Chemotherapy targets include the following: (1) bone marrow–derived proangiogenic cells that adhere to the walls of new blood vessels and further stimulate their growth by paracrine mechanisms, (2) circulating/cycling ECs present in sprouting blood vessel capillaries, and (3) authentic bone marrow–derived circulating endothelial progenitor cells that can incorporate into the lumen of growing vessels and differentiate into ECs. Chemotherapy also suppresses or reduces the secretion of soluble tumor-derived growth factors and soluble endothelial growth factors, which is the key component of angiogenesis.
Because multiple signaling pathways are involved in angiogenesis and more than one pathway is often dysregulated in human tumors, blocking a single pathway may not be highly effective and/or can lead to resistance when the tumor cells develop other angiogenesis mechanisms. By combining antiangiogenic agents with each other or with other modalities used in the treatment of cancer, the limitations of each therapeutic approach may be overcome, leading to enhanced efficacy with diminished toxicity and potentially overcome or delay drug resistance.
In PCa, a unique systemic therapy for advanced PCa is ADT. ADT is initiated in patients who develop metastatic disease or in patients receiving radiation therapy. In mice bearing LNCaP tumors, castration resulted in a rapid decrease in VEGF mRNA expression as well as markedly reduced tumor neovascularization. This finding is also true in vitro and in vivo in another androgen-responsive cell line CWR22Rv1. The reversal of neovascularization was also demonstrated after hormone withdrawal as an early event in human PCa response to ADT. It was discussed previously that androgen activates the HIF-1 signaling pathway in androgen-sensitive LNCaP cells. HIF-1 helps maintain survival for PCa cells in an androgen-deprivation environment. EGF and other growth factors are mediators of this crosstalk. Agents inhibiting the HIF-1 signaling pathway can potentially sensitize PCa to ADT and are the logical choice for combining them with AR signaling inhibitors.
In this review, the authors focus on the key combination studies performed in the past several years in the treatment of PCa and hope to understand more about the complex antiangiogenic strategies for PCa treatment and shed light for future trial designs.
Related posts:
 Targeting the Androgen Receptor
Targeting the Androgen Receptor
 Moving Toward Personalized Medicine in Castration-Resistant Prostate Cancer
Moving Toward Personalized Medicine in Castration-Resistant Prostate Cancer
 Palliative Care in Castrate-Resistant Prostate Cancer
Advanced Clinical States in Prostate Cancer
The Experience with Cytotoxic Chemotherapy in Metastatic Castration-Resistant Prostate Cancer
Palliative Care in Castrate-Resistant Prostate Cancer
Advanced Clinical States in Prostate Cancer
The Experience with Cytotoxic Chemotherapy in Metastatic Castration-Resistant Prostate Cancer
 Management of Docetaxel Failures in Metastatic Castrate-Resistant Prostate Cancer
Management of Docetaxel Failures in Metastatic Castrate-Resistant Prostate Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


