Fig. 11.1
The pathogenesis of lupus nephritis and novel therapies based on pathogenic pathways. In this model lupus nephritis begins with the accumulation of autoreactive self-antigens such as nucleosomes. The nucleosomes accumulate because of deficient clearance or overwhelming production and can drive anti-dsDNA production (Step 1). The resulting IC accumulate in the renal vascular beds (Step 2), in part because nucleosomes contain positively charged histones. After deposition, IC can activate the complement system, activate circulatory leukocytes via expressed FcγR, and activate resident cells expressing TLRs (Step 3). This establishes a cascade of inflammatory cytokine and chemokine production that recruits and activates inflammatory cells, lymphocytes, and pDCs. These infiltrating cells further amplify the intrarenal production of cytokines and chemokines. The result is a locally driven and accelerated autoimmune response with Th1 characteristics and increased IC accumulation and accompanying complement and FcγR activation. This response culminates in the production of inflammatory mediators of tissue damage (Step 4). One immediate consequence is interruption of the glomerular filtration barrier through damage to glomerular endothelial cells (EC), glomerular basement membrane (GBM), and podocytes (P), which leads to proteinuria and hematuria, the clinical hallmarks active lupus nephritis. Throughout this model, novel therapies are shown that are being tested, have been tested, or are anticipated to be tested in clinical trials of SLE and LN. These therapies are related to the pathogenic pathways, and their specific targets along these pathways are indicated. Note: CR complement receptor, MAC complement membrane attack complex
The Genetics of LN
Considerable progress has been made toward identifying the genetic basis of SLE, using genome-wide and candidate gene studies (reviewed in [14, 15]). This has resulted in the identification of over 30 genes that appear to be related to specific pathogenic pathways in SLE, including IC clearance/inflammatory pathway genes, immune response genes, and interferon alpha (IFN-α) signaling and response genes. Unfortunately, the same effort has not yet been applied to the genetics of human LN [16]. Furthermore, the studies that have been done suggest considerable heterogeneity of LN genetic susceptibility in differential racial/ethnic groups, implying that a complete picture of LN genetics will not emerge until studies are repeated in all at-risk populations [16]. For example, genome studies have identified six quantitative trait loci (QTLs) that are linked to LN [17, 18], three regions are linked in European Americans, and three are linked in African Americans. Despite these caveats, some genes bear further consideration [19] and are discussed below.
Fc receptors for IgG (known as Fcγ receptors, or FcγR) are engaged by IC and can provide either protection from IC-mediated injury by facilitating the phagocytosis and clearance of IC or induce inflammatory responses by activating the cells expressing FcγR (reviewed in [20, 21]). Studies of polymorphic forms of FcγR have clarified which role dominates in SLE. There are three classes of FcγR (FcγRI, FcγRII, and FcγRIII), with different genes that produce full-length products for FcγRII (FcγRIIA, FcγRIIB, FcγRIIC) and FcγRIII (FcγRIIIA and FcγRIIIB). Single nucleotide polymorphisms (SNPs) that affect the peptide sequence have been identified in some of these genes that influence binding affinity for IgG, including the FcγRIIA 491G > A SNP (changing amino acid 131 from arginine to histidine) and the FcγRIIIA 559T > G SNP (changing amino acid 158 from phenylalanine to valine) [22–24]. Though not unequivocal, most studies have reported that the lower affinity forms of FcγRIIa (R131) and FcγRIIIa (F158) are associated with SLE and particularly with LN [25, 26]. The fact that the forms of these receptors that bind IC more efficiently are associated with protection against SLE suggests that their overall function is to promote IC clearance rather than drive tissue inflammation and that relative deficiencies in this function contribute to LN.
Two cytokines important for cell infiltration into the kidney, monocyte chemotactic protein-1 (MCP-1) and interleukin (IL)-18, have promoter polymorphisms that influence expression levels. MCP-1 attracts monocytes and T cells, and IL-18 attracts plasmacytoid dendritic cells (pDCs). The MCP-1 variant that results in higher expression levels is associated with LN [27]. Similarly, the IL-18 variant that causes higher expression is associated with diffuse proliferative LN [28].
Signal transducer and activator of transcription-4 (STAT4) is important for transmitting interferon (IFN)-αsignals. STAT4 has a genetic variant that is associated with increased STAT4 RNA levels and with SLE, particularly LN [29, 30].
Autoantibodies and Immune Complexes
Autoantibodies are synonymous with lupus, especially antinuclear and anti-double-stranded (ds)DNA antibodies [33, 34]. Over 100 self-antigens have been identified in SLE patients that are targets of autoantibodies, including dsDNA, single-stranded (ss)DNA, nucleoproteins, RNA–protein complexes, ribosomes, phospholipids, carbohydrates, cell cytoplasm and cell surface molecules, blood components, and endothelial cells [35]. Because autoantibodies to all of these antigens are not present in every patient, autoantibody patterns may predispose to specific organ involvement. In this regard autoantibodies to dsDNA and complement component C1q seem to be particularly relevant to LN.
Several studies have found an association of high titer anti-dsDNA with active LN (reviewed in [36]). Furthermore, anti-dsDNA antibodies can be isolated from the glomeruli of LN patients [37–39]. This may be due to the nature of the dsDNA antigen in the context of SLE. Nucleosomes are composed of DNA in association with a core of positively charged histone proteins, are released by cells undergoing apoptosis, and can be trapped in the glomeruli, perhaps facilitated by electrostatic interaction between positively charged histones and the negatively charged glomerular basement membrane [40]. Anti-dsDNA recognizes DNA in nucleosomes, and the binding of anti-dsDNA in lupus renal tissue occurs at the site of glomerular nucleosome deposition [41]. In addition, there appears to be cross-reactivity between anti-dsDNA and antigens in kidney tissue [42], in particular alpha-actinin expressed on podocytes and mesangial cells [43], and annexin II on mesangial cells [44]. Regardless of the predominant mechanism, the result is localized anti-dsDNA-containing IC with the potential to drive local tissue inflammation. Anti-dsDNA autoantibodies appear to be mainly IgG1 and IgG3 [45, 46], which are the most inflammatory IgG subtypes due to their ability to activate complement and engage Fc receptors for IgG.
Antibodies to C1q, the first component of the classical complement pathway, have been strongly associated with LN in many but not all studies [47–50]. Anti-C1q does not appear to cause an acquired deficiency of circulating C1q because anti-C1q binding requires a neoepitope formed when C1q becomes fixed to its target substrate. Rather, injury is likely related to interaction of anti-C1q with C1q already present in the kidney, such as in IC bound to nucleosomes [51, 52]. The resulting C1q/anti-C1q IC could focus an inflammatory response to the kidney, similar to anti-dsDNA, leading to nephritis. However, unlike anti-dsDNA antibodies, most anti-C1q antibodies appear to be IgG subclass 2 [53, 54], which is a poor complement activator and low-affinity antigen for Fc receptors. Other IgG subtypes (mainly IgG1) can be present in these IC, so the role of anti-C1q in LN pathogenesis may depend on the relative amounts of each anti-C1q IgG subtype.
There is some evidence suggesting that glomerular accumulation of autoantibodies and IC might not be necessary for the development of LN. An MRL/lpr mouse model of lupus with a mutant transgene that prevented B cell secretion of immunoglobulin developed interstitial nephritis and vasculitis that was similar to the wild-type MRL/lpr [55]. However, the glomerular component of the disease consisted mainly of focal glomerular atrophy and not the typical glomerulonephritis that develops in wild-type MRL/lpr. Other studies have suggested that IC are not sufficient for the full development of LN and that T cells are also required. Subsets of T cells appear to significantly contribute to the progression of LN in the NZB/NZW mouse model of lupus, including activated CD4 T cells [56] and Th17 T cells [57]. Although caution must be taken when extrapolating these results to human LN, the role IL-17 may be particularly relevant, as discussed below.
The Complement System
The formation of IC leads to the activation of the complement cascade which can provide protective effects against SLE, mainly by promoting proper clearance of circulating IC. However, once IC are deposited in tissue, complement can drive tissue inflammation and damage, either through direct effects on tissue (complement membrane attack complex) or by activating cells to produce pro-inflammatory cytokines and toxic mediators.
Complement may provide protection from SLE by solubilizing IC, so they are less likely to become trapped in tissue [58, 59], clearing apoptotic debris, an immunogenic source of self-antigens, through opsonization by C1q [60, 61], and clearing IC through C4b/C3b/C3bi receptors after opsonization by the complement activation products C4b and C3b/bi [62, 63]. The type one complement receptor (CR1, CD35) binds C4b, C3b, and C3bi, is expressed in the circulation predominantly on erythrocytes (E-CR1), and mediates the binding of complement-opsonized IC to erythrocytes (a process known as immune adherence) [64]. This binding allows erythrocytes to shuttle IC through the circulation, minimizing glomerular trapping of IC and promoting IC delivery to the liver and spleen for safe removal [64]. The evidence that all of these complement functions protect against SLE includes studies showing that individuals with homozygous deficiencies of classical pathway components have an increased risk for developing SLE and SLE-like diseases [65, 66] and that E-CR1 levels are decreased in SLE and fluctuate in chronically active disease [67–69].
In contrast, there is substantial circumstantial evidence that complement-mediated inflammation and complement-mediated direct tissue damage contribute to the pathogenesis of LN: (1) Circulating levels of C3 and C4 are lower in active LN compared to inactive LN or nonrenal SLE, indicating ongoing complement activation [70–73]. A longitudinal assessment of circulating C3 and C4 levels during SLE flare showed a decrease at renal flare but not at nonrenal flare, even if the nonrenal flare occurred in patients with a history of LN [69]. (2) Complement components, including the membrane attack complex, C3, and factor B, are deposited or produced in LN kidneys [70, 74–80]. (3) The inflammatory receptors for C3a and C5a are expressed or upregulated in the glomerular endothelium, mesangium, and podocytes of LN kidneys [81, 82]. (4) The expression of regulators of complement activation, including CR1 and decay accelerating factor (CD55), is decreased in LN kidneys [83–87].
The fact that complement can both protect against the development of SLE and yet drive LN speaks to the complex and multifaceted functions of the complement system. It also suggests that the overarching role of complement in SLE pathogenesis is initially to contribute protection against SLE onset. However, once the disease is established, with IC accumulation in the tissue, the role of complement is reversed to that of a significant contributor to disease progression, especially as it involves the kidney.
Renal Chemokines and Cytokines
Complement activation in the kidney due to IC and IC themselves initiate further intrarenal inflammation that becomes the hallmark of kidney injury in LN. One consequence of complement activation is the generation of the membrane attack complex, which damages cell membranes through the formation of transmembrane pores [76]. Another consequence of complement activation and also Fc receptor activation by IC is the induction of pro-inflammatory chemokine and cytokine expression by renal parenchymal cells and resident leukocytes [88]. These chemokines and cytokines recruit inflammatory leukocytes to the kidney, which in turn accelerate inflammation through secretion of additional chemokines and inflammatory cytokines and, in the case of neutrophils and monocytes, induce tissue damage through release of proteolytic enzymes and pro-oxidants. Examples of upregulated chemokines and cytokines in kidneys of LN patients include MCP-1, macrophage inflammatory protein-1-alpha (MIP-1α), IL-6, IL-10, IL-12, IL-17, IL-18, IFN-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), and Eta-1/osteopontin [88–95]. Deletion or inhibition of the expression of these cytokines in experimental models of SLE and LN significantly attenuates kidney injury [96–100]. In human LN intervention studies, targeting some of these cytokines is being done and if successful will verify their role in the pathogenesis of human LN.
Recent evidence is particularly strong for IL-17 playing an important role in the pathogenesis of LN. The two major cell sources of IL-17 in SLE/LN are Th17 cells and CD4−CD8− T cells, and these have been observed in renal biopsies of LN patients [95]. Local production of IL-17 may drive inflammatory cytokine and chemokine expression by resident glomerular and tubular cells having the IL-17 receptor [101], leading to activation of infiltrating neutrophils and monocytes [102, 103]. The presence of IL-17-producing cells in the LN kidney may also represent a shift away from natural regulatory T cells capable of suppressing immune responses (see below) [104].
T and B Cells
The cytokine profile of infiltrating renal T cells suggests their role in LN. Intrarenal production of Th1 cytokines, specifically IL-12, IFN-γ, and IL-18, appears to exceed Th2 cytokines in proliferative LN and correlates with histologic activity [90, 93, 94, 105]. The Th2 cytokine IL-10 does increase in LN, but overall the Th1/Th2 ratio is higher. Th1-dominant expression can also be observed in serum, urine, and circulating T cells of LN patients [105, 106]. Th1 responses are associated with activated macrophages and with the production of IgG capable of activating complement and FcγR pathways, all implicated in the pathogenesis of kidney injury. The Th1 dominance displayed in LN patients, both locally in the kidney and systemically in the circulation, suggests that this may be an important prerequisite for developing LN. It should be noted that, while the above studies indicate a shift to a predominant Th1 profile in LN, a recent report suggests that IgE, which is a consequence of a Th2 response, is an important component of LN development [107].
Human regulatory T cells (Treg), characterized as CD4+CD25hiFoxP3+, inhibit immune responses through effects on T and B cells and particularly autoantibody production [108–111]. In experimental animals there is an inverse correlation between circulating Treg numbers and circulating anti-dsDNA levels [112], and lupus-like activity, including glomerulonephritis, can be suppressed by adoptive transfer of Tregs [113, 114]. Human SLE studies generally also show fewer circulating Tregs [109, 115], but the implication of this loss of regulatory T cells for LN remains unclear [116, 117].
Infiltrating B cells have also been described in human LN kidneys. Their presence may directly target autoantibodies to the kidney, as has been shown in NZB/NZW mice [118]. B cells in renal tissue may also present kidney antigens to intrarenal T cells. Recent work has shown that intrarenal B and T cells associate with various degrees of organization, including structures resembling germinal centers with central follicular dendritic cells [119, 120]. Interestingly, these structures appear to occur mainly outside of the glomeruli and are associated with tubular basement membrane IC [120]. These may contribute specifically to tubulointerstitial inflammation in LN.
Interferon-Α and Plasmacytoid Dendritic Cells
Recent work from several laboratories indicates a central role for IFN-α in the pathogenesis of SLE and LN [117]. IFN-α is produced mainly in response to nonself-stimuli, such as microorganisms. For example, it can be induced by single-stranded viral RNA and unmethylated, nonmammalian DNA [121, 122]. Although a number of cell types can produce IFN-α, pDCs appear to be a major source, and production is induced after stimulation of their intracellular Toll-like receptors 7 and 9 (TLR 7, 9) among others [121–123]. Another major source appears to be the recently described low-density granulocytes (LDGs) [124]. In addition to potentially secreting pathophysiological amounts of IFN-α, LDGs also express abnormal levels of neutrophil extracellular traps (NETs), containing autoantigens and IL-17, and induce further synthesis of IFN-α, by pDCs [125]. The effects of IFN-α on the immune response include driving maturation of conventional dendritic cells into potent antigen-presenting cells [126], inducing B cell differentiation to plasma cells [127], and contributing to the development of CD4 helper T cells [128] and CD8 memory T cells [129].
Important for SLE, TLR 7 and 9 can engage mammalian nucleic acids in the context of ICs [130, 131]. The autoantibody component of the IC allows pDC to internalize the IC via FcγRIIa, thereby bringing the nucleic acids in proximity to TLR 7 and 9 [132]. IFN-α generated in this way may facilitate an immune response that contributes to breaking tolerance to self-nucleic acids. This paradigm implies a preexisting presence of autoantibodies in patients destined to develop SLE, and in fact ANA are found in >1 % of the general population [133–135].
Supporting a role for IFN-α in SLE is the observation that patients treated with IFN-α can develop a lupus-like illness [136–140]. Furthermore, there is an increase in IFN-α-induced gene expression, known as the IFN-α signature, in many patients with active SLE [141, 142]. With respect to LN, it has been shown that during severe LN, pDC disappear from the circulation and accumulate in glomeruli, due in part to glomerular expression of IL-18 and pDC expression of the IL-18 receptor [143, 144]. It is plausible that glomerular IC containing dsDNA (e.g., nucleosomes) could drive these pDCs to produce IFN-α, amplifying the intrarenal autoimmune response and contributing to the formation of local germinal centers. Additionally, peripheral blood cell levels of IFN-α-inducible genes are associated with LN patients [142, 145], and IFN-α-inducible chemokines, including MCP-1, are associated with active LN and LN flare [146–148]. Studies in many [149] but not all [150] mouse models also support a role for IFN-α in LN pathogenesis.
The apparent importance of the IFN-α pathway in SLE pathogenesis has reinvigorated the concept that microbial pathogens may be environmental factors contributing to the initiation of SLE. The activation of TLRs that stimulate IFN-α by viral and bacterial nucleic acids may be important in breaking tolerance or in accelerating the autoimmune response.
Diagnosis of Lupus Nephritis
Most patients with LN do not present with overt signs of kidney disease but rather abnormalities of serum creatinine and/or the urine, so kidney involvement must be specifically sought. Forty to sixty percent of patients who eventually have clinical renal involvement have findings of kidney disease at the initial diagnosis of lupus [5, 6, 9]. Therefore, at a minimum, patients should be assessed for LN during the initial evaluation for SLE, whenever there is suspicion of a flare of SLE activity, and if otherwise stable, at least yearly. The rationale for close monitoring is that preservation of kidney function in patients with LN is best achieved with early diagnosis and treatment [151–157].
There are some caveats in using serum creatinine as a screening tool for LN in the lupus population. A creatinine in the normal range of the clinical laboratory may be abnormally high for a woman with small-moderate muscle mass and low rates of creatinine production. In this circumstance comparison to previous creatinine measurements would be ideal, but these are not always available. In addition, hypoalbuminemic patients with severe nephrotic syndrome may have increased tubular creatinine secretion, lowering serum creatinine, and leading to an impression of better renal function than in actuality [158]. Finally, SLE patients may develop acute renal insufficiency and an increase in serum creatinine because of infection, medications, nephrotoxins, hemolysis, thrombosis, and cardiac failure. These conditions should be excluded in order to attribute a rise in serum creatinine to LN.
A urine dipstick positive for blood and/or protein in a patient with SLE is suggestive of LN; however, a systematic study of the accuracy of the urine dipstick as a screening tool found a false-negative rate in up to 30 % of SLE patients and a false-positive rate in about 40 % of patients [159]. Therefore, the urine sediment should be evaluated for evidence of glomerulonephritis. Glomerular bleeding is suggested by acanthocytes and/or red blood cell casts. White blood cells and white blood cell casts in the absence of infection are indicative of renal inflammation and consistent with a diagnosis of glomerulonephritis.
Proteinuria is a key indicator of kidney injury in SLE and is used as a clinical biomarker of relapse, remission, and successful treatment. It also has pathogenic importance because proteinuria may injure the kidney. Therefore, accurate measurement of protein excretion is crucial to the ongoing monitoring and management of LN.
Random spot urine protein-to-creatinine (P/C) ratios can be used in addition to urine dipsticks to screen patients for proteinuria but are not accurate enough to be used to make therapeutic decisions or to follow changes in proteinuria magnitude in response to therapy. The most reliable method to quantify proteinuria is to measure the P/C ratio of a 24-h urine collection or an intended 24-h collection that is at least 50 % complete [160]. Measuring the P/C ratio reduces confounding the assessment of proteinuria by errors in collecting the 24-h urine. A 12-h overnight urine collection that includes the first morning void urine also provides an accurate measure of proteinuria magnitude and may be easier for patients to collect [161].
The Kidney Biopsy and Renal Pathology in SLE
A kidney biopsy is the gold standard for the exact diagnosis and classification of LN, but LN is not a renal biopsy diagnosis. The pathologic changes described below are characteristic, but not diagnostic of LN unless the patient also fulfills the American College of Rheumatology criteria for SLE. In the absence of a concurrent clinical diagnosis of SLE, only a diagnosis of immune complex glomerulonephritis can be made, with the suggestion that the glomerulonephritis could be associated with SLE.
A biopsy is not necessarily required if the only clinical abnormalities suggesting LN are isolated hematuria or minor proteinuria in the absence of hematuria and an active urine sediment. A biopsy should be considered when proteinuria is above 500 mg/day, because this degree of proteinuria has been associated with significant kidney injury [162–168].
The first kidney biopsy of a patient with suspected LN, while important diagnostically and therapeutically, has limited prognostic value because most of the active lesions are reversible with treatment. However, a follow-up biopsy performed after several months of treatment may provide important prognostic information in terms of progression to or lack of chronic lesions [169–172]. In addition to determining prognosis, repeat biopsies may help to decide if and how therapy should be changed if the clinical response is other than a complete remission. Thus, several groups are beginning to suggest more liberal use of the kidney biopsy in the ongoing management of patients with LN.
The clinical utility of the kidney biopsy depends on obtaining an adequate sample of renal cortex (at least 10 glomeruli) and examination by a renal pathologist [173]. In as much as every biopsy is a clinical-pathological correlation, the nephropathologist should be given all relevant clinical information in order to properly interpret the tissue and integrate the microscopic findings with the clinical data. Furthermore, it is essential that the clinician and pathologist review the findings together before initiation of therapy to ensure that specific clinical concerns are addressed and that the lesions have been appropriately contextualized.
Classification Schemes for LN
LN is currently classified by the International Society of Nephrology (ISN)/Renal Pathology Society (RPS) criteria developed in 2003 [174]. Similar to the previous WHO classification of LN, the ISN/RPS classification is based primarily on light microscopic patterns of glomerular injury. A criticism of both systems is the lack of consideration of the tubulointerstitium.
The ISN/RPS classification (Fig. 11.2) differentiates active (A) from chronic (C) and segmental (S) from global (G) glomerular lesions. Active glomerular lesions include glomerular endocapillary hypercellularity with or without leukocyte infiltration and with reduction of capillary lumina, karyorrhexis, fibrinoid necrosis, rupture of the glomerular basement membrane, cellular or fibrocellular crescents, wire loop lesions, and large intraluminal immune complexes (hyaline thrombi). Chronic glomerular lesions include glomerular sclerosis (segmental or global), fibrous adhesions, and fibrous crescents. Segmental lesions involve less than half of the glomerular capillary tuft area; global lesions involve more than 50 % of the glomerular capillary tuft area. The definition of segmental and global lesions is somewhat controversial. The degree of involvement in a given tissue section depends on the plane of the section through the glomerular tuft. Thus, depending on the level of the cut, a segmental lesion could appear to involve more or less than 50 % of the glomerular capillary surface area. Nonetheless, with these rules ISN/RPS classifies kidney injury in SLE as follows:
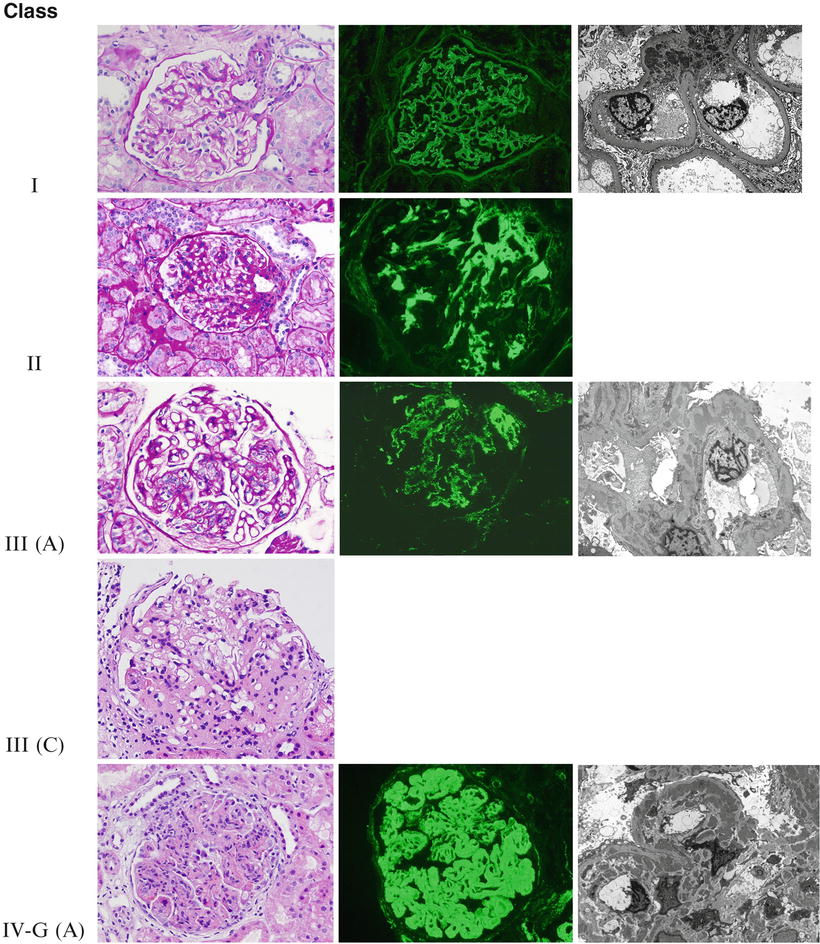
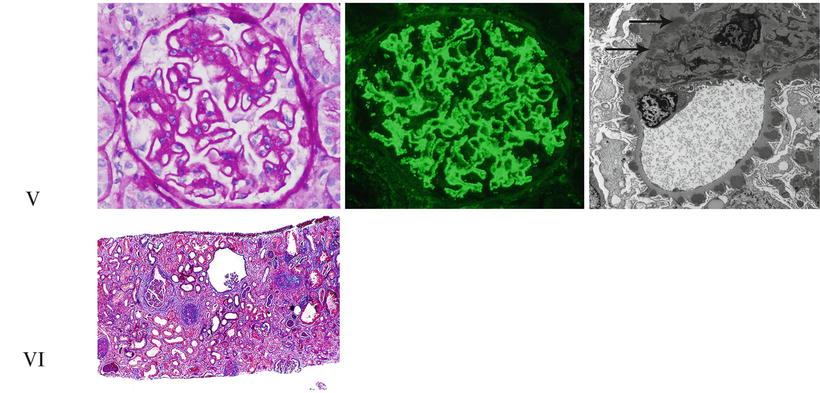
Fig. 11.2
The ISN/RPS classification of lupus nephritis. Representative photographs of the different lupus nephritis classes as seen under light, immunofluorescence, and electron microscopy. Class I: Light microscopy shows a normal-appearing glomerulus in Class I lupus nephritis (PAS ×200). Immunofluorescence demonstrates fine granular paramesangial IgG deposits (direct immunofluorescence ×400), and electron microscopy shows mesangial electron-dense immune-type deposits (uranyl acetate, lead citrate ×5,000). Class II: Mild mesangial expansion and hypercellularity are seen on light microscopy in Class II lupus nephritis (PAS ×200), while immunofluorescence shows mesangial IgG deposits (direct immunofluorescence ×400). Electron microscopy will be similar to Class I. Class III (A): Segmental glomerular intracapillary hypercellularity by light microscopy in focal proliferative lupus nephritis (PAS ×200). Granular mesangial and segmental glomerular capillary staining for C1q by direct immunofluorescence (×400). Several intramembranous, subendothelial, and occasional subepithelial deposits are demonstrated along the glomerular capillary loops by electron microscopy (uranyl acetate, lead citrate ×8,000). Class III (C): On light microscopy, a segmentally sclerosing glomerulus shows chronicity in focal proliferative lupus nephritis (H&E ×200). Class IV-G (A): Light microscopy shows a globally hypercellular glomerulus with segmental necrosis and apoptotic debris in a case of active diffuse proliferative lupus nephritis (H&E ×200). Immunofluorescence shows prominent widespread glomerular capillary and mesangial staining for C1q (direct immunofluorescence ×400). There are abundant large subendothelial, mesangial, and occasional subepithelial electron-dense immune-type deposits in active diffuse proliferative lupus nephritis (uranyl acetate, lead citrate ×5,000). Class V: Membranous lupus nephritis, like idiopathic membranous nephropathy, shows diffusely thickened glomerular capillary loops (PAS ×200). Direct immunofluorescence with an antibody to IgG reveals widespread granular glomerular capillary and mesangial staining (×400). Abundant subepithelial electron-dense deposits are present, often with mesangial electron-dense deposits (arrows; uranyl acetate, lead citrate ×8,000). Class VI: Advanced sclerosing lupus nephritis shows widespread interstitial fibrosis (blue) and mostly globally sclerotic glomeruli (Masson’s trichrome ×40)
Class I: Minimal Mesangial LN
Glomeruli appear normal by light microscopy, but immunofluorescence and electron microscopy reveal mesangial immune complex deposits.
Clinical pathologic correlation: Usually no clinical kidney abnormalities; often normal serum complement.
Class II: Mesangial Proliferative LN
In additional to mesangial immune complexes as in Class I, there is pure mesangial hypercellularity without glomerular endocapillary hypercellularity or crescents.
Clinical pathologic correlation: Normal kidney function, mild hematuria and/or proteinuria, often normal serum complement.
Class III: Focal LN
Endocapillary and/or extracapillary (crescents) proliferative lesions are seen in fewer than 50 % of glomeruli. Glomerular lesions in focal LN are almost always segmental. By immunofluorescence and electron microscopy, mesangial immune complexes are seen, usually with segmental glomerular capillary immune complex deposits. Class III (A) shows only active lesions (focal proliferative LN).
Class III (A/C) has both active and chronic lesions (focal proliferative and sclerosing LN). In such cases, focal or segmental sclerosing glomeruli coexist with glomeruli with active proliferative/necrotizing lesions. Class III (C) shows only focal sclerosing glomerular lesions (focal sclerosing LN). Active lesions are not seen.
Clinical pathologic correlation: Normal or impaired kidney function, nephritic sediment, proteinuria (may be nephrotic), often low serum complement.
Class IV: Diffuse LN
Endocapillary and/or extracapillary (crescents) glomerular proliferative lesions are seen in more than 50 % of glomeruli. Glomerular lesions can be global or segmental, and active or chronic glomerular lesions are evaluated separately. Immunofluorescence and electron microscopy show mesangial and capillary loop immune complex deposits. The glomerular capillary loop deposits are mainly subendothelial and often large. Class IV-S(A) or G(A) indicates active diffuse segmental or global endocapillary or extracapillary proliferative glomerular lesions involving more than 50 % of the glomeruli. Class IV-S(A/C) or G(A/C) indicates segmental or global diffuse proliferative and sclerosing LN. In such biopsies, active proliferative lesions coexist with chronic sclerosing glomerular lesions.
Class IV-S(C) or G(C) indicates diffuse segmental or global sclerosing LN. In these subclasses, no active lesions are present, only inactive sclerosis or scarring.
Clinical pathologic correlation: Normal or impaired kidney function, nephritic sediment, proteinuria (may be nephrotic), often low serum complement.
It has been suggested that the pathogenesis of LN with true global lesions is different from LN with segmental glomerular lesions and that this affects outcomes and treatment choices [175–179]. Class IV LN with lesions involving more than 50 % of the glomerular tuft area (classified as Class IV-G) appears to have a worse outcome than global proliferative LN with 100 % involvement of the glomerular capillary tuft area (also classified IV-G by ISN/RPS) and Class IV-S with less than 50 % glomerular tuft involvement [177]. However, other studies did not find any difference in outcome between patients with Class IV-S and Class IV-G LN [180–184], but this may be because cases of Class IV-G with lesions involving more than 50 % of the glomerular tuft were generally not separated out from Class IV-G with 100 % tuft involvement [177–179]. At the present time, these concerns remain unresolved.
Class V: Membranous LN
Glomeruli do not reveal endocapillary hypercellularity; the mesangium may be normocellular or hypercellular. As in idiopathic membranous glomerulonephritis, spike formation on methenamine silver stain is common. Glomerular subepithelial immune complex deposits involve over 50 % of the glomerular capillary tufts in over 50 % of the glomeruli, and in contrast to idiopathic membranous nephropathy, mesangial immune complexes are almost invariably present. Also different than idiopathic membranous glomerulonephritis, in Class V LN the IgG immune deposits contain mainly IgG1 and IGg3 as opposed to IgG2 and IgG4 [185]. Class V LN is common in combination with Class III or Class IV LN. In these combined patterns of injury, the proliferative component is listed first (such as Classes III + V or Classes IV + V).
Clinical pathologic correlation: Normal kidney function, often nephrotic syndrome, microscopic hematuria, often normal serum complement.
Class VI: Advanced Sclerosing LN
Over 90 % of the glomeruli are globally sclerosed without residual activity. There has to be clinical or morphologic evidence that the advanced glomerular sclerosis is secondary to LN. Immunofluorescence and electron microscopy still frequently reveal mild glomerular immune complex deposits in the few non-sclerotic glomeruli.
Clinical Pathologic Correlation: Chronic kidney disease.
The ISN/RPS classification, while clinically useful [184, 186, 187], is still based purely on morphologic findings and arbitrary definitions. Kidney biopsies can and should be exploited in novel ways to better inform the understanding of LN pathogenesis and the development of new therapeutics. For example, leukocyte subsets can be analyzed by specific staining in lupus kidneys and may yield new insights on renal inflammation [188]. Proteomic techniques can be used to look for patterns of protein expression in LN [189, 190]. Gene expression in biopsies can be analyzed with microarray techniques [89, 190]. As these technologies are applied to kidney biopsies, they will undoubtedly enhance the amount of information available from renal tissue and result in a new molecular/functional classification of LN kidney biopsies.
Class Transformation in LN
Follow-up biopsies of LN often show a class different from the initial biopsy [172, 191]. Successful treatment of Class III or IV may result in improvement to Class II LN, whereas treatment failures may end in Class VI. Class I or II LN may evolve to any of the higher classes. Another common transformation is for Class III to become Class IV. Class III or IV may transform into Class V LN or Class V into Class III or IV. In these cases the resulting LN often shows a proliferative plus membranous pattern.
Activity and Chronicity Indices in LN
In an effort to standardize the evaluation of LN kidney biopsies, a scoring system was developed for active and chronic lesions [192–194]. The scoring criteria are described in Table 11.1. While the prognostic and therapeutic value of these activity and chronicity indices is debated [195], the score gives the clinician a readily understood estimate of the severity and acuity of the LN.
Table 11.1
Activity and chronicity indices for lupus nephritis biopsies
Scorea | Crescents | Indicators of activity | Indicators of chronicity | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Glomerular necrosis/karyorrhexis | Glomerular neutrophils | Endocapillary hypercellularity | Large subendothelial immune deposits | Interstitial inflammation | Glomerular sclerosis | Fibrous crescents | Tubular atrophy | Interstitial fibrosis | ||
None | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Mild | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
Mod | 4 | 4 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
Sev | 6 | 6 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
Immunofluorescence Findings in LN
Classically, glomerular immune complex deposits in LN often show a “full house” immunofluorescence pattern, meaning that all or almost all immunoreactants (IgG, IgA, IgM, kappa and lambda light chains, C1q, C3) are present. This is unusual in other forms of glomerulonephritis. Importantly, the absence of full house immunofluorescence does not exclude LN, especially in Class V. C1q staining is usually quite prominent in LN and rare in other forms of glomerulonephritis. Another characteristic immunofluorescence feature in LN biopsies is the frequent deposition of immune complexes along the tubular basement membrane, the peritubular capillary basement membrane, Bowman’s capsule, and arterial and arteriolar walls. Most LN immune complexes contain IgG1 and IgG3, less IgG2, and minimal IgG4. Interestingly glomerular and extraglomerular immune complexes frequently have different IgG subclass distribution [196], suggesting different mechanisms for the deposition of glomerular and extraglomerular immune complexes.
Electron Microscopy in LN
In addition to immune complex deposits (discrete electron-dense immune-type deposits), a very common ultrastructural finding in LN is the tubuloreticular inclusion (Fig. 11.3). Tubuloreticular inclusions are seen mainly in endothelial cells and, while not diagnostic of LN, reflect high interferon levels in patients with active SLE. They are not restricted to renal endothelial cells but can be found throughout the body.
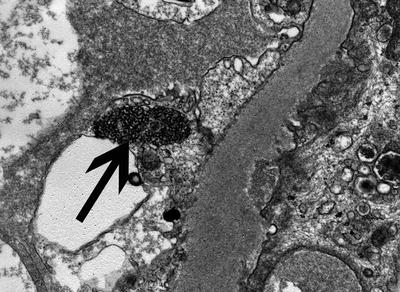
Fig. 11.3
A large tubulorecticular inclusion in a glomerular capillary endothelial cell (arrow). Such inclusions are common in all cases of LN and reflect increased interferon levels (Uranyl acetate, lead citrate, ×20,000)
Tubulointerstitial Lesions in LN
Light microscopic lesions in the tubulointerstitium are nonspecific. Interstitial inflammatory cell infiltrates may or may not be present in biopsies with LN. They are more common in patients with proliferative LN (Class III or IV) and indicate an active disease process. Interestingly, the degree of interstitial inflammatory cell infiltrate does not correlate with the degree of tubulointerstitial immune complex deposition [196, 197]. In later stages of LN, interstitial fibrosis and tubular atrophy appear and indicate progressive chronic injury. Interstitial nephritis in the absence of LN may also occur [198]. Interstitial fibrosis and tubular atrophy may or may not be associated with active inflammatory cell infiltrate in the same biopsy specimen.
Other Kidney Lesions in Patients with SLE
Not all kidney disease in SLE patients is the classic, immune-complex-mediated glomerulonephritis known as lupus nephritis. Several non-LN glomerular diseases have been reported in SLE patients [198–203]. This literature is mostly case reports, but in a series of 252 patients, 5 % were found to have changes consistent with focal segmental glomerulosclerosis, minimal change disease, thin glomerular basement membrane disease, hypertensive nephrosclerosis, and amyloidosis [200]. The incidence of podocytopathies in lupus patients appears to be greater than in the general population, suggesting a causal link to the immune dysregulation of SLE [204, 205]. AA amyloidosis has also been reported in some series [198, 201–203].
Thrombotic microangiopathy (TMA) with or without LN is not uncommon, particularly in patients who have circulating antiphospholipid antibodies and high d-dimer levels [206–208]. The biopsy findings (Fig. 11.4) include arterial/arteriolar fibrin thrombi with or without fibrinoid necrosis of the vessel wall, fragmented red blood cells in the fibrin thrombi or embedded in the thickened loosened arterial/arteriolar walls, and mucoid subendothelial widening of the arteries/arterioles. In more chronic stages, concentric thickening (onion skinning) of the arterial/arteriolar walls may develop. Arterial/arteriolar immune complex deposits may or may not be present. The glomerular changes include fibrin thrombi and/or prominent thickening of the glomerular capillaries, secondary to subendothelial electron lucent widening between the glomerular capillary basement membrane and the swollen endothelium (seen on electron microscopy). Because of the capillary wall thickening, the glomerular capillary lumen is narrowed and many of these glomeruli appear “bloodless.” Fragmented red blood cells are not unusual in the glomerular capillaries.
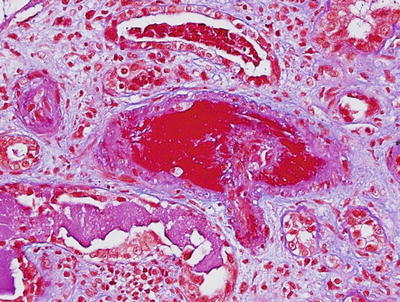
Fig. 11.4
A thrombus occluding an interlobular artery in a patient with lupus nephritis and antiphospholipid antibodies (Masson’s trichrome ×200)
Management of Lupus Nephritis
Recently three separate guidelines for the management of LN have been published. The guidelines have come from the American College of Rheumatology [209], the Kidney Disease-Improving Global Outcomes work group [210], and the Joint European League Against Rheumatism and the European Renal Association-European Dialysis and Transplant Association [211]. These groups were not totally independent, because some of the lupus experts worked with more than one of the sponsoring organizations. The major recommendations of all of the guidelines and of this chapter are evidence based and fairly consistent. A commentary comparing the LN guidelines and puts them into perspective is also available [212].
Proliferative and membranous forms of LN generally require treatment with immunosuppressive agents. However, because these therapies have considerable toxicity, their use should be informed by clinical and pathologic severity. For example, in advanced stage sclerosing Class III (C) or Class IV (C), the therapeutic strategy should shift from a focus on immunosuppression, except as needed for extrarenal SLE, to a focus on renal protection. Renal protection is also used for Class VI LN. The goal of renoprotection in inactive sclerosing LN is to prolong kidney function and avoid ESRD requiring renal replacement therapy for as long as possible. Renoprotective strategies include control of blood pressure with antiproteinuric antagonists of the renin-angiotensin-aldosterone system, sodium restriction, protein restriction, and correction of metabolic abnormalities and are discussed elsewhere in this text.
In addition to immunosuppression, all SLE patients should receive hydroxychloroquine unless they have a specific contraindication. Antimalarials have activity against TLR7 and 9, and as discussed previously TLRs are important in the pathogenesis of SLE and LN [213–215].
Hydroxychloroquine may protect against vascular thrombosis [216], kidney damage [217], renal flares [218], and ESRD [219] and has a favorable impact on lipid profiles.
Mesangial Lupus Nephritis
Class I LN is rarely diagnosed because there are no or few clinical renal manifestations that would warrant a kidney biopsy. Patients with Class II LN may have glomerular hematuria and proteinuria (usually non-nephrotic), but kidney function is normal [220, 221]. The immunomodulatory regimens used to treat extrarenal SLE are generally sufficient for Class II (and I), along with renoprotective measures for hypertension and proteinuria as clinically indicated. If nephrotic proteinuria is present, a podocytopathy such as minimal change disease or focal segmental glomerulosclerosis should be considered and, if found, treated as for the idiopathic variants of these glomerular diseases [200, 204, 205].
Proliferative Lupus Nephritis
Proliferative LN (Class III or IV) can be an aggressive disease that requires intense therapy. A general strategy for the treatment of proliferative LN is outlined in Fig. 11.5. All current approaches initiate treatment with a cytotoxic agent and a corticosteroid. This is done because pioneering randomized clinical trials conducted by the National Institutes of Health (NIH) showed that although corticosteroids were effective in controlling proliferative LN, adding a cytotoxic agent at the beginning of treatment decreased the frequency of renal relapse and the development of future CKD or ESRD [222, 223]. Importantly, the beneficial effect of cytotoxic agents to preserve kidney function was only apparent after 3–5 years of follow-up [222–224].
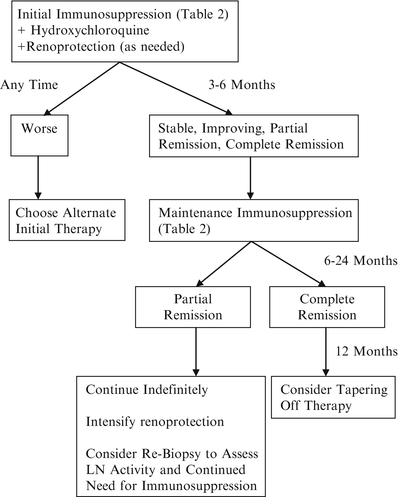
Fig. 11.5
A general strategy for the treatment of proliferative lupus nephritis
Based on the early NIH trials, proliferative LN was treated with the cytotoxic agent cyclophosphamide for 18 months or more, a regimen associated with considerable morbidity. Limiting cyclophosphamide to 6 months however resulted in an increase in renal relapses [223]. The need for therapy beyond 6 months was thus apparent, but cyclophosphamide seemed too toxic. This problem was addressed in a prospective study of azathioprine (AZA), mycophenolate mofetil (MMF), or intravenous cyclophosphamide after 6 months of initial cyclophosphamide therapy [225]. Over 72 months patients treated with AZA or MMF were significantly less likely to reach the composite end point of death or CKD than the cyclophosphamide-only group and experienced fewer adverse side effects. Thus, the treatment strategy for proliferative LN evolved into an induction phase of high-dose corticosteroids plus cyclophosphamide for 6 months, followed by substitution of an antimetabolite, usually AZA or MMF for cyclophosphamide, for a prolonged maintenance phase. Although induction could imply a remission is achieved after induction, this is not generally the case with LN (see below). Initial phase may be a more appropriate description. Initial therapies for proliferative LN are given in Table 11.2.
Table 11.2
Treatment options for proliferative LN
Initial therapya | Dose | Duration |
|---|---|---|
Cyclophosphamide-modified NIH | 0.5–1 g/m2 IV monthly | 6 months |
Cyclophosphamide-oral | 1–1.5 mg/kg/dayb | 2–4 monthsc |
Cyclophosphamide-Euro-lupus | 500 mg IV q2 weeks | 3 months |
MMFd | 1–3 g/day | 6 months |
Maintenance therapy | Dose | Duration |
MMFd | 1–2 g/day | 1 year beyond complete remission Indefinitely in partial remission |
AZA | 1.5–2.5 mg/kg/day | 1 year beyond complete remission Indefinitely in partial remission |
CSA | 2.5–3 mg/kg/day | 1 year beyond complete remission Indefinitely in partial remission |
The most widely used cyclophosphamide delivery format for initial LN treatment is intravenous pulses of 0.5–1.0 g/m2 given monthly for 6–7 pulses. Oral cyclophosphamide shows comparable efficacy to intravenous cyclophosphamide, is easier to administer, and generally costs less [222, 226–231]. Oral cyclophosphamide lost favor because several studies associated it with increased toxicity, especially cystitis [222], but many of these early studies used very high doses (up to 2.5 mg/kg/day) for 6 or more months. Lower-dose, shorter-duration oral cyclophosphamide (Table 11.2) is effective, well tolerated, and results in a cumulative cyclophosphamide exposure similar to 6 months of pulse therapy [232]. In an effort to reduce cyclophosphamide exposure in LN, a true low-dose cyclophosphamide induction regimen (Table 11.2) was compared to 6 monthly pulses followed by two quarterly pulses of cyclophosphamide [233, 234]. This low-dose regimen was termed “Euro-lupus,” and after 10 years of follow-up, the end points of death, ESRD, and doubling of the serum creatinine were similar in both groups, suggesting that low-dose cyclophosphamide can be used successfully in proliferative LN. Importantly, the Euro-lupus patient population was mostly Caucasian, and the proliferative LN was of mild-moderate severity.
The major morbidities with any cyclophosphamide regimen are infertility, future malignancy, and infection. To protect fertility women may be offered prophylaxis with leuprolide and men testosterone while cyclophosphamide is being given [235, 236]. Sperm banking and ovarian tissue cryopreservation are additional options. To decrease the risk of future malignancy, lifetime cumulative exposure to cyclophosphamide should be <36 g [237, 238]. To reduce infectious morbidity, the white blood cell count should be monitored weekly and the dose adjusted to keep the neutrophil count ≥2,000 cells/μ[mu]l. In patients with moderate-severe kidney insufficiency, reduce the cyclophosphamide dose by 20–30 % [239].
To completely eliminate the undesirable side effects of cyclophosphamide, The Aspreva Lupus Management Study (ALMS) prospectively compared MMF + corticosteroids to intravenous pulse cyclophosphamide + corticosteroids, looking for superiority in response at the end of a 6 month induction period [240]. This end point was not achieved; however, the ALMS induction trial showed an equivalent response to MMF and cyclophosphamide at 6 months with a similar incidence of adverse events, serious infections, and deaths for each drug. In a post hoc stratification of ALMS results by race and ethnicity, black or mixed-race patients who received intravenous cyclophosphamide did not do as well as those who received MMF, and the response rate among Hispanic patients was greater with MMF [241]. These findings suggest that Black and Hispanic patients, generally considered to have more resistant LN [242], may respond better to MMF than intravenous cyclophosphamide, but this will need to be verified in an independent prospective trial.
The ALMS study MMF target dose was 3 g/day. An increasing number of studies indicate that MMF dose and mycophenolic acid (active metabolite) levels do not correlate well, but that levels do influence therapeutic response and toxicity [243–245]. Therapeutic drug monitoring for MMF may thus prove useful in managing LN patients, especially if patients are not responding as expected and to avoid over treatment.
AZA was also prospectively compared to cyclophosphamide and showed a similar clinical outcome, but repeat biopsy after treatment showed more chronic damage in the AZA group, and those treated with AZA had a higher incidence of renal relapse and doubling of the serum creatinine [246, 247]. Because of this AZA was not placed as an initial therapy in Table 11.2, but in some areas of the world, AZA may be the only option due to cost or availability, and at least some large retrospective studies have shown long-term responses similar to initial treatment with cyclophosphamide [248].
The calcineurin inhibitors tacrolimus (TAC) and cyclosporine A (CSA) have recently been tested as an alternative to cyclophosphamide for initial therapy in proliferative and mixed proliferative plus membranous LN [249, 250]. Although these trials suggest similar complete and partial remission rates in the short term, the number of patients enrolled was small, and prospective long-term follow-up is lacking. The role for calcineurin inhibitors in treating proliferative LN remains to be determined in long-term prospective randomized trials.
Leflunomide is currently used to treat rheumatoid arthritis and works by blocking lymphocyte proliferation, T cell activation, and suppressing production of cytokines such as interleukin-2. Response rates were similar to those of cyclophosphamide when leflunomide was used to treat LN in two small trials from China [251, 252]. Repeat kidney biopsies at 6 months showed a large increase in the chronicity index in one of these studies [251], but this was not seen in repeat biopsies from the second study [252]. Thus, long-term trials will be required to determine if leflunomide preserves kidney function over time as well as cyclophosphamide.
As will be discussed later, the overall response rates for Class III and IV LN with any of the initial therapies are only about 60 % at 6–12 months, with fewer complete remissions. With plenty of room for improvement, the anti-B cell monoclonal antibody rituximab was added onto MMF and corticosteroids in a randomized controlled trial to determine if 12-month outcomes could be improved [253]. This large trial was based on several small, open-label, uncontrolled trials that suggested rituximab may be effective in proliferative LN, either for refractory disease or as initial therapy [254–260]. However, at 12 months there were no differences between the rituximab and placebo groups in terms of complete or partial remissions. Thus, rituximab cannot be recommended as adjunctive initial therapy.
Therefore, for most patients with proliferative LN, the current choice for induction/initial therapy is between a cyclophosphamide-containing regimen and an MMF-only regimen. A number of factors must be considered in making this choice. Some investigators favor a full-dose (modified NIH or oral) cyclophosphamide-based regimen for patients with very severe LN because many patients in the two largest studies of MMF versus cyclophosphamide had less severe LN than the patients in some of the randomized clinical trials of cyclophosphamide [224, 240, 261, 262]. In contrast, others point out that in the ALMS trial, the subset of patients with severe LN did as well with either drug (presented at an American Society of Nephrology Meeting). MMF should be considered if patients have a cumulative dose of cyclophosphamide approaching 36 g. Low-dose (Euro-lupus) cyclophosphamide could be considered in Caucasians with moderate LN.
One more variable needs to be factored into treatment choice, and that is long-term preservation of kidney function. As mentioned previously, the long-term benefit of cytotoxic therapy was seen only after 3–5 years of follow-up [222–224]. There are a few studies comparing the long-term benefit of initial MMF versus initial cyclophosphamide. In a Chinese cohort, there were no significant differences in renal function between groups after a median of 64 months [231]. However, more MMF-treated patients had relapses, prolonged proteinuria >1 g/day, and persistent serum creatinine >2 mg/dL, all variables associated with deterioration of kidney function over time. The ALMS Maintenance Trial (discussed below) was a 3-year extension of the ALMS trial designed to evaluate maintenance regimens [263]. Although not designed to compare the long-term efficacy of initial therapy on kidney function, there was a (nonsignificant) trend toward fewer treatment failures in those who received cyclophosphamide as initial therapy as opposed to MMF, and this was independent of the choice of maintenance therapy. Thus, it cannot yet be stated with certainty that initial therapy of proliferative LN with MMF is equivalent to cyclophosphamide in preserving kidney function.
Several factors may be used to assess how patients are doing during initial therapy. If patients become clearly worse during the first few weeks to months of therapy, with continued loss of kidney function, increasing proteinuria, or increasing activity of the urine sediment, moving them to an alternate initial treatment protocol seems reasonable (Fig. 11.5). Post hoc analysis of the ALMS data showed that a positive renal response at 6 months could be predicted at 8 weeks if patients experienced a reduction in proteinuria of ≥25 % or a normalization of complement components C3 and/or C4 if they had low complement levels at the beginning of treatment [264]. This was not dependent on whether the patient was in the MMF or cyclophosphamide arm. The positive predictive value of these variables was about 70 %, and therefore they could be helpful in guiding treatment changes. Nonetheless, few patients reach complete remission by 6 months. In six studies of Black, White, and Hispanic patients [225, 232, 233, 240, 246, 261] treated with intravenous cyclophosphamide (modified NIH), low-dose cyclophosphamide (Euro-lupus), MMF, or AZA, the median complete plus partial remission rate 6 months after starting treatment was 54 % (range 18–85 %), and the complete remission rate was only 8.6 % (range 7.4–25 %). The median complete plus partial response rate 12 months after therapy was 60.5 % (range 32–85 %). In contrast, in four Chinese cohorts treated with MMF or cyclophosphamide [227–230], the median complete response rate at 12–24 months was 71 % (range 57–81 %), with an overall response rate of 90 % (range 73–95 %). Clinical improvement in Class III/IV LN continues well beyond 6 months and into the maintenance phase of therapy [229, 232, 233, 246, 265]. Consistent with this, kidney biopsies after 6 months of initial therapy generally show improvement in inflammation but rarely complete resolution of pathologic changes [49, 188, 266, 267]. In summary one may expect approximately half of LN patients to achieve a complete or partial response by 12 months and another 5–25 % to respond by 24 months. Considering only complete responses, half are achieved by 12 months and the other half by 20–24 months.
Maintenance therapies after the initial treatment of proliferative LN are outlined in Table 11.2. AZA and MMF are the most commonly used drugs for maintenance. To determine if AZA or MMF is superior, the ALMS trial was extended for 3 years in patients who achieved a complete or partial remission after the 6-month induction phase [263]. These patients were re-randomized to maintenance with MMF or AZA and followed prospectively. Over 3 years the composite treatment failure end point (death, ESRD, renal flare, sustained doubling of serum creatinine, or requirement for rescue therapy) was reached in 16 % of maintenance MMF-treated patients compared to 32 % of maintenance AZA-treated patients (P = 0.003). The superiority of MMF over AZA was not dependent on initial therapy (MMF or cyclophosphamide) or race of the patient. In contrast, the smaller MAINTAIN trial prospectively compared MMF to AZA as maintenance therapy in a predominantly Caucasian population after initial treatment with the low-dose Euro-lupus cyclophosphamide protocol, regardless of whether patients had achieved remission [268]. The primary end point was time to renal relapse, and after at least 3 years of follow-up, MMF and AZA were found to be statistically equivalent, although MMF was numerically better. These studies suggest MMF may be the first line for maintenance therapy, but the choice must be individualized to patient-specific factors such as desire for pregnancy and side effects.
CSA may also be considered for maintenance therapy. A pilot randomized clinical trial in 69 patients with Class III/IV LN suggested that 2 years of CSA may be as effective as 2 years of AZA for maintenance after initial treatment with prednisone and oral cyclophosphamide [269]. The outcomes measured in this study were relapse and proteinuria. Another randomized clinical trial showed CSA was as effective as AZA in terms of tapering maintenance corticosteroids in severe systemic lupus, but only 29 % of the patients had LN [270].
There is little evidence to guide the duration or withdrawal of maintenance therapy, but long treatment times are commonly used. For example, immunosuppression was continued for an average of 3.5 years in seven randomized clinical trials [222, 223, 225, 228, 231, 233, 234, 246]. Also supporting the need for a prolonged maintenance phase, only 40 % of proliferative LN patients showed biopsy evidence of improvement to Class II after 2 years of intense immunosuppression [247]. After complete remission is achieved and has been sustained for at least a year, or longer in patients with a history of renal flares, it is reasonable to consider slowly tapering therapy. Unfortunately there is no standard definition of complete remission for LN in the literature. Nonetheless, controlling proteinuria is important for preserving kidney function, and the target for complete remission should be a proteinuria level less than 0.5 g/day [271, 272]. Serum creatinine should improve to a patient’s pre-LN baseline if known, although serum creatinine may be increased (acceptably) by renoprotective therapies. Thus, at a minimum serum creatinine should remain stable. Urine sediment should not have any RBC or WBC casts, but hematuria may persist for months [273]. Although reassuring, resolution of abnormal serologies is not necessary for remission of LN [274–280].
For patients who achieve only a partial remission, defined generally as a stable or improved serum creatinine and a reduction of proteinuria ≥50 % and to below nephrotic-range, immunosuppression should be continued indefinitely, and renoprotective therapies intensified (Fig. 11.5). Kidney biopsies done 2 or more years after initial treatment often still show activity in the presence of continued significant proteinuria and/or abnormal serum creatinine [281, 282]. Increasing corticosteroids or using alternative immunosuppressive agents to convert a partial to complete remission is not supported by evidence. In partial responders consideration should be given to repeat biopsy to determine the level of pathologic activity. Significant activity may provide a rationale for re-induction therapy, while significant sclerosis/fibrosis may provide a rationale for tapering immunosuppression other than what is needed for extrarenal SLE.
The long-term treatment objectives in proliferative LN are to prevent renal flares, ESRD, and death. Examining several studies that included Black, Hispanic, Caucasian, and Chinese patients observed for a median of 6 years (range 3–10 years), mortality and ESRD were 5 % (range 0–20 %) and 4 % (range 0–10 %), respectively [225, 227, 229, 246, 247]. Doubling of serum creatinine occurred in 7.2 % (range 0.04–18.2 %) of patients and renal flare in 23 % (range 0.04–42 %). Similarly, during 10 years of follow-up after treatment with low-dose (Euro-lupus) cyclophosphamide, 25 % of patients reached the composite end point of death, doubling of serum creatinine, or ESRD [234].
After diagnosis of proliferative LN, the question of predicting how a patient will do in the short and long term often comes up. This has been addressed in several prospective and retrospective studies, using univariate and multivariate analyses. As expected, multivariate analyses demonstrated that many of the factors identified in univariate analyses were not truly independent. A summary of independent risk factors for LN outcomes is shown in Table 11.3 [227–229, 242, 247, 265, 266, 283, 284]. From these data, two observations suggest that biomarkers currently used to predict outcomes are inadequate. First, almost none of the biomarkers are consistently identified across studies looking at the same outcome. Second, several studies identify no independent risk factors for many of the outcomes. This may be explained by study design, patient populations, variations in operational outcome definitions, and/or the strength of the current selection of biomarkers. An exception is the initial serum creatinine, which was identified across studies and outcomes as a biomarker of future remission, renal relapse, chronic kidney disease, and ESRD. It is interesting that failure to achieve a complete remission was identified by only a few investigations to be a significant risk factor for chronic kidney disease, ESRD, or mortality [157, 229, 285]; in most proteinuric kidney diseases, resolution of proteinuria is the strongest predictor of renal survival [286–288].
Table 11.3
Risk factors for outcomes in proliferative LN
Variablea | Hazard ratio or relative risk | Retrospective studyb | Prospective study |
|---|---|---|---|
Ability to achieve remission | |||
White race | 2.63 | × | |
Class IV LN | 2.05 | × | |
Proteinuria | 0.86 for every g/day increase | × | |
Therapy delayed >3 months | 0.58 | ||
Serum Crc | 0.21 for every mg/dL increase | × | |
Serum Cr | 0.96 for every μ[mu]mol/L | × | |
Noned | × | ||
Renal flare | |||
Class IV LN | 0.28 | × | |
Time to remission | 1.03 per month | × | |
CNS lupus | 8.41 | × | |
Chronicity indexe | 1.22 per point | × | |
Persistently + DS-DNA | 2.94 | × | |
Activity indexf | 1.13 per point | × | |
CRg not achieved | 6.2 | × | |
None | × | ||
Doubling serum Cr | |||
Serum Cr > 1 mg/dL | 4.1 | × | |
Hispanic | 3.6 | × | |
Povertyh | 3.5 | × | |
Govt insurancei | 3.0 | × | |
Nephritic flare | 17.7 | × | |
Chronicity index | 2.1 per point | × | |
None | × | ||
ESRD | |||
Serum Cr | 2.8 for every mg/dL increase | × | |
No remission | 6.8 | × | |
Anti-Ro positive | 2.35 | × | |
Class III LN ≥ 50 %j | 2.77 | × | |
None | × | ||
Composite: Doubling serum Cr, ESRD, death | |||
Serum Cr | 1.26 for every mg/dL increase | × | |
Chronicity index | 1.18 per point | × | |
Composite: ESRD, death | |||
Age > 50 | 3.3 | × | |
Non-White race | 2.28 | × | |
Serum Cr | 2.32 | × | |
Membranous Lupus Nephritis
Membranous LN (Class V) is a nonproliferative glomerulopathy that can be seen in 8–20 % of LN patients [289–291]. While less aggressive than proliferative LN, 20 % of Class V patients may develop CKD over time, and ESRD develops in about 8–12 % of patients [289–292]. In addition, left unchecked, the nephrotic proteinuria of Class V LN predisposes to hyperlipidemia, atherosclerosis, and thrombotic events [3, 287, 293]. Unlike idiopathic membranous nephropathy, spontaneous remission of heavy proteinuria occurs in only a minority of Class V LN patients [294, 295]. For these reasons Class V LN, especially with heavy proteinuria, does warrant therapy.
Renoprotective and antiproteinuric therapies should be used for pure membranous LN with low-level proteinuria. Class V LN patients with nephrotic-range proteinuria and/or renal insufficiency should be considered for immunosuppression. In contrast to proliferative LN, there has only been one prospective, randomized clinical trial for Class V [296]. This showed that the addition of cyclophosphamide (six intravenous pulses of 0.5–1 g/m2 every other month) or CSA (5 mg/kg/day for 11 months) to corticosteroids improved the 12-month complete plus partial response rate from 26 % to 60–83 %, respectively. However, 40 % of the CSA group relapsed within a year of finishing treatment, but relapses were not seen in the cyclophosphamide group for 48 months. A subgroup analysis of Class V patients from the ALMS trial concluded that MMF (2–3 g/day) and cyclophosphamide were equally efficacious for Class V [297]. Consistent with this a number of small, nonrandomized, retrospective, or open-label studies also showed that MMF and AZA (1–2 mg/kg/day) are effective in Class V LN [298–301], achieving a total remission rate of 40–60 % within 4–6 months [300, 302].
Because kidney function is not likely to deteriorate rapidly in Class V, it is reasonable to try to induce remission with MMF or AZA (plus corticosteroids) and switch to the potentially more toxic cyclophosphamide or CSA only if that fails. Failure to achieve remission is predicted by an initial proteinuria level over 5 g/day [296]. Failure to achieve sustained remission is a risk factor for decline in kidney function in Class V, independent of race or ethnicity [296].
Patients with mixed membranous and proliferative LN are generally treated as for the proliferative component but may have a less favorable prognosis [290]. Alternatively, in a small randomized, controlled trial from China in patients with mixed Class IV and V LN, the combination of TAC (4 mg/day), MMF (1 g/day), and oral corticosteroids (multi-target therapy) was compared to pulse monthly IV cyclophosphamide (0.75 g/m2 for 6 months) plus oral corticosteroids. At 6 months 90 % of patients treated with the “multi-target” therapy achieved either complete or partial remission compared to 45 % of patients treated with cyclophosphamide (P = 0.002) [303].
Follow-Up of the LN Patient
LN is a characteristically relapsing disease. A survey of LN patients who had participated in randomized clinical trials showed flares in 40 % of complete responders within a median of 41 months of remission and 63 % of partial responders within a median of 11.5 months of response [304]. Renal flare risk factors were discussed previously (Table 11.3), but there is little consensus on what predisposes to a flare. Kidney biopsies done after successful treatment often show an increase in the chronicity index, suggesting that each flare can cause some chronic damage that may culminate in CKD or ESRD [49, 169, 170, 247, 266, 267, 303, 305].
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


