Stress-ulcer bleeding in critically ill patients develops as a result of various risk factors, notably prolonged mechanical ventilation and coagulopathy. The incidence of clinically significant bleeding is decreasing secondary to widespread use of potent antisecretory medications and improved intensive care. This article describes the epidemiology, pathogenesis, risk factors, and management of stressrelated bleeding. The various prophylactic agents should be used judiciously to prevent unwanted drug side effects. Standard algorithms for critically ill patients are needed to identify those at high risk and to delineate criteria for the use of prophylactic therapeutic options in these patients.
Stress is defined as a response to severe demands on the human body resulting in the disruption of homeostasis through physical and psychological stimuli. It has long been recognized that severe physiologic stress can cause gastric mucosal damage. More than 150 years ago, Curling described duodenal ulcer bleeding in patients with extensive burn injuries. The syndrome of stress-related mucosal damage (SRMD) of the gastrointestinal (GI) tract was first described in 1971 by Lucas and colleagues who used the term stress-related erosive syndrome . Since then, numerous terms have been used to describe stress-related mucosal damage in critically ill patients, including stress ulcers, stress erosions, stress gastritis, hemorrhagic gastritis, erosive gastritis, and stress-related mucosal disease .
Epidemiology
Acute GI bleeding in critically ill patients has been described in the medical literature since the 1800s. Determining the true frequency of clinically important bleeding is complicated by variability in the definitions of end points, difficulty in measuring the end points, and the heterogeneity of the patient populations. The reported incidence of stress-related mucosal damage varies from 6% to 100% in critically ill patients. Endoscopic studies generally indicate that approximately 75% to 100% of critically ill patients have gross gastric lesions visible when endoscopy is performed within the first 1 to 3 days of illness. The prevalence of stress-related mucosal damage ranges from 15% to 50% if occult bleeding (defined as a drop in hemoglobin level or positive stool occult blood test) is used as an end point. Clinically overt bleeding (hematemesis or nasogastric lavage positive for bright red blood) occurs in about 5% to 25% of critically ill patients who do not receive prophylactic therapy. However, clinically overt bleeding does not predict impending clinically significant bleeding (defined as bleeding associated with hypotension, tachycardia, and a drop in hemoglobin level necessitating transfusion). Approximately 20% of clinically evident bleeding is reported to be clinically significant. The incidence of clinically significant GI bleeding has been estimated to be approximately 3% to 4%.
In a prospective study of more than 2000 patients, Cook and colleagues reported an incidence of clinically significant bleeding of 1.5% in critically ill patients. Clinically important or significant bleeding was defined as overt bleeding complicated by one of the following within 24 hours after the onset of bleeding (in the absence of other causes): (1) a spontaneous decrease of more than 20 mm Hg in the systolic blood pressure; (2) an increase in heart rate of more than 20 beats per minute; or (3) a decrease in systolic blood pressure, measured on sitting up, of more than 10 mm Hg; or (4) a decrease in hemoglobin level of more than 2 g/dL and the need for subsequent transfusion, after which the hemoglobin did not increase by a value defined as the number of units transfused minus 2 g/dL. The mortality rate was 48.5% in the group with bleeding and 9.1% in the group without bleeding ( P <.001). Another recent prospective study examined the attributable mortality and length of intensive care unit (ICU) stay in relation to clinically important GI bleeding in more than 1500 patients. Using the matched cohort method, the relative risk (RR; the ratio of the probability of the event occurring in the exposed group versus a nonexposed group) of mortality attributable to clinically significant GI bleeding was significant (RR 2.9, 95% confidence interval [CI] 1.6 to 5.5). The mortality attributable to bleeding was also statistically significant using the model-based matched cohort method (RR 1.8, 95% CI 1.1 to 2.9). The study also tested the risk of mortality attributable to bleeding over time, using the unadjusted regression method. Patients who bled earlier in their ICU stay had a lower risk of dying than patients who bled later ( P = .02); the RR of mortality associated with clinically important bleeding was 0.4 (95% CI 0.06 to 2.0) at 2 weeks, 1.6 (95% CI 0.6 to 4.0) at 3 weeks, and 7.4 (95% CI 1.7 to 32.2) at 4 weeks. After adjusting for age, APACHE II score, admitting diagnosis, ventilation status, and 6 Multiple Organ Dysfunction Score (MODS) domains, the ICU stay attributable to bleeding by the regression method was 6.2 days (95% CI 1.0 to 11.4 days).
To summarize, the reported incidence of clinically significant bleeding is approximately 1.5% to 4% with the relative risk of mortality at two to four. The attributable length of ICU stay associated with clinically significant bleeding is approximated to five to seven days. However, the incidence of bleeding from stress-related mucosal damage seems to be decreasing secondary to therapeutic advancement and prevention of mucosal hypoperfusion.
Pathogenesis
The gastric mucosa maintains structural integrity and function despite continuous exposure to noxious factors, including 0.1 mol/L HCl and pepsin, which are capable of digesting tissue. An early hypothesis proposed by Hunter in 1772 and supported by Virchow in 1853 was that continuous circulation of alkaline blood through the mucosa neutralizes acid. Subsequent work demonstrated that a large number of mucosal defense mechanisms prevent mucosal damage and maintain mucosal integrity. Vane discovered that the major mechanism by which aspirin and other nonsteroidal antiinflammatory drugs (NSAIDs) produce gastric damage involves inhibition of prostaglandin (PG) synthesis. The concept of cytoprotection developed in the late 1970s and early 1980s by Robert and colleagues sparked tremendous interest in gastric mucosal defense mechanisms. Gastric mucosal injury may occur if noxious factors “overwhelm” an intact mucosal defense or the mucosal defensive mechanisms are impaired.
Gastric Mucosal Defense Mechanisms
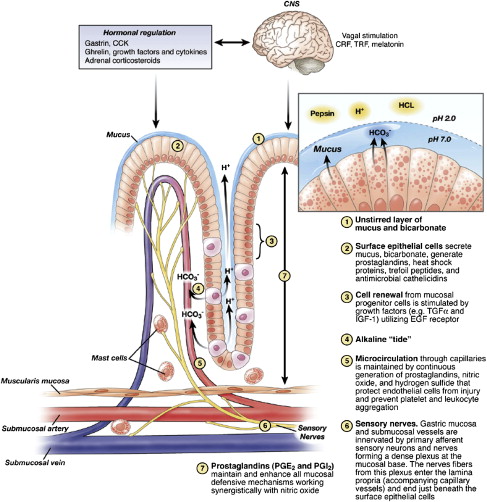
Defense mechanisms permit the gastric mucosa to withstand frequent exposure to damaging factors. These include local mechanisms such as the mucus-bicarbonate-phospholipid barrier, continuous cell renewal from mucosal progenitor cells, alkaline tide, and mucosal microcirculation. These concepts have been recently reviewed and illustrated by Laine and colleagues. Fig. 1 depicts the key components of gastric mucosal defense. Various peptides, including gastrin 17, cholecystokinin, thyrotropin-releasing hormone, bombesin, corticotropin-releasing factor (CRF), epidermal growth factor (EGF), peptide YY, neurokinin A analogs, and intragastric peptone, exert gastroprotection, which is abolished by afferent nerve denervation, blockade of calcitonin gene-related peptide receptors, and inhibition of NO synthase.
Ischemia and Reperfusion Injury
Stress-related gastric mucosal injury seems to be related to local ischemia, although progression to significant mucosal injury also requires acid. Critically ill patients developing acute gastric mucosal lesions show a significant decrease in gastric mucosal blood volume compared with controls. Studies in the rat model revealed a significant correlation between blood pressure during hypotension and gastric mucosal blood flow. Mucosal blood flows measured by the hydrogen gas clearance technique in the corpus, antrum, and duodenum of rats all showed a significant linear correlation with mean blood pressure. Blood flow to the mucosa in the stomach and duodenum decreased progressively as systemic blood pressure fell. Significant gastric mucosal lesions occurred only after mean blood pressure and, hence, mucosal blood flow was reduced to less than 40% of baseline values. A rat model of hemorrhagic shock and retransfusion-induced gastric injury revealed that histologic mucosal damage occurred even without acid, although it increased with intragastric instillation of acid in a dose-dependent fashion. In addition, reperfusion after ischemia increased histologic injury compared with ischemia alone, although increasing the duration of ischemia (from 20 to 30 minutes) increased gastric histologic lesions to the same extent as a similar period of retransfusion (10 minutes after 20 minutes ischemia). In the absence of acid, gross gastric lesions were minimal, involving 4% of the body and 3% of the antrum surface area. However, intragastric instillation of acid (0.1 mol/L HCl) markedly increased the gross lesions in a dose-dependent fashion to 53% and 45% of the body and antral surface areas, respectively. These studies imply that splanchnic hypoperfusion in the presence of gastric acid leads to stress-related mucosal damage and suppression of gastric acid would minimize such injuries. This forms the basis for the use of gastric antisecretory agents as prophylactic agents for stress-related mucosal injuries.
Oxidative Stress
Under hypoxic-ischemic conditions, reactive oxygen species (ROS), such as superoxide anions, hydrogen peroxide, and hydroxyl radicals, are rapidly and continuously produced. Oxidative stress resulting from hypoxia and ischemia to the gastric and duodenal mucosa has proved to be an important element in the development and progression of epithelial necrosis and mucosal ulceration. Overproduction of ROS results in oxidative damage, including lipid peroxidation, protein oxidation, and DNA damage, which can lead to cell death. Furthermore, ROS are known to act as second messengers to activate diverse redox-sensitive signaling transduction cascades, including mitogen-activated protein kinases (MAPKs) and downstream transcription factors such as NF-κB and AP-1. These signaling pathways and transcription factors regulate the expression of numerous pro-inflammatory genes and, thereby, lead to the elaboration of chemical and humoral mediators of tissue inflammation and injury. The p38 MAPK is considered important in determining cellular adaptive responses and cell fate. It has been demonstrated that ROS-mediated p38 activation plays an essential role in the pathogenesis of stress-induced gastric inflammatory damage in the rat model of cold immobilization stress. These results suggest that local ROS generation could be an initiating or triggering event in the early phase of stress-induced gastric mucosal injury. The clinically relevant translation of this area of research is the importance of restoring and maintaining systemic and regional circulation and tissue oxygenation in critically ill patients.
The Cyclooxygenase-2 Pathway
Whereas cyclooxygenase-2 (COX-2) has no essential role in the maintenance of gastric mucosal integrity under basal conditions, COX-2 is rapidly induced in a pro-ulcerogenic setting and contributes to mucosal defense by minimizing injury. Ischemia/reperfusion has been shown to increase COX-2 messenger RNA levels in the rat gastric mucosa. It has also been discovered that hypoxia increases expression of the COX-2 gene in human vascular endothelial cells in culture, independent of other stimuli. Therefore, COX-2 expression is assumed to be up-regulated as one of the protective mechanisms when the stomach is exposed to stress, and contributes to mucosal defense by minimizing injury. Therefore, it is reasonable to speculate that COX-2 may prevent gastric mucosal ulcers in mild stress by a phenomenon of adaptive cytoprotection, but may not be able to prevent stress-related mucosal damage in severe stress because of the intensity of the insult and activation of various other pro-ulcerogenic mechanisms. It is understandable why NSAID exposure in the critically ill patient may have profound adverse effects on sustaining the COX-2 defense mechanism.
Endogenous Nitric Oxide and Endothelin-1
Gastric microcirculatory disturbances are involved in the pathogenesis of stress ulcers; however, the vasomodulators causing this vascular instability are not fully understood. Reduced local mucosal nitric oxide (NO; a vasodilator) generation and increased endothelin-1 (a potent vasoconstrictor) levels seem to be involved in events associated with compromise of mucosal blood flow observed in SRMD. In addition, NO plays an important role in the regulation of epithelial cell functions in the GI tract. Gene expression of inducible NO synthase (iNOS) is markedly up-regulated following ischemia/reperfusion and is accompanied by a significant increase in the mucosal NO content. Endogenous NO plays a dual role in ischemia/reperfusion-induced gastric injury; constitutive NO synthase/NO is protective, whereas inducible NO synthase/NO can be pro-ulcerogenic during ischemia/reperfusion. The constitutive NO synthase/NO acts to maintain mucosal integrity through modulating various functions such as mucus secretion, bicarbonate secretion, and mucosal blood flow. In contrast, the inflamed mucosa is associated with recruitment of various inflammatory cells as well as up-regulation of iNOS, cytokines, and adhesion molecules. Under stress conditions, NO produced by iNOS reacts with O 2 − produced by neutrophils to form peroxynitrites. These NO-derived reactive compounds contribute to the nitrosative and oxidative stress burden of vulnerable tissue such as the mucosa of the stomach and duodenum. Thus, the duality of NO function is dependent on the vascular and metabolic integrity of the mucosal and submucosal tissues and the presence or absence of inflammation. Similarly, endothelin-1 levels in plasma and injured mucosa of critically ill patients have been found to be elevated, suggesting endogenous ET-1 may play an important role in the local pathogenesis of stress ulcers.
Mucosal Blood Flow
Critically ill patients may experience a decrease in gastric mucosal blood flow through systemic hypotension and splanchnic hypoperfusion. In rats, gastric lesions began to appear when blood pressure fell to 33% of baseline during hemorrhagic shock. At blood pressure 80% below baseline, 26.8% of the total corpus area developed lesions. Normotensive critically ill septic patients on mechanical ventilation were reported to have a 50% to 60% reduction in gastric mucosal blood flow, measured by reflectance spectrophotometry, compared with controls. Fig. 2 illustrates the consequences of mechanical ventilation in critically ill patients. Splanchnic hypoperfusion in mechanically ventilated patients is caused by decreased cardiac output, release of pro-inflammatory cytokines, and sympathetic system activation. Mechanical ventilation as a risk factor for stress-related mucosal damage is discussed further in next section.
In summary, the etiology and pathophysiology of SRMD is multifactorial and, although several steps in this process have been elucidated, there are additional questions that require further investigation. Ischemia and reperfusion are believed to cause disruption of the mucosal defenses, resulting in mucosal injury and ulceration. The major causative factors of SRMD that have been recognized to date include reduced mucosal blood flow, ischemia, and reperfusion injury. The integrity of the gastric mucosa is maintained by several factors, including the microcirculation and the mucus-bicarbonate gel layer. Maintenance of these functional and structural elements provides nutrient and oxygen delivery to the epithelial cells, neutralization of hydrogen ions to prevent acid back diffusion, and dissipation of reactive substances and other toxic cellular byproducts. Thus, stress-related mucosal damage develops if these elements of protection are either compromised or breached. Tissue PG levels have been shown to be decreased, which in turn impairs mucus replenishment and microcirculation responsiveness. Elevated levels of nitric oxide synthase have also been shown to contribute to reperfusion injury and necrotic cell death, and tissue inflammation response.
Pathogenesis
The gastric mucosa maintains structural integrity and function despite continuous exposure to noxious factors, including 0.1 mol/L HCl and pepsin, which are capable of digesting tissue. An early hypothesis proposed by Hunter in 1772 and supported by Virchow in 1853 was that continuous circulation of alkaline blood through the mucosa neutralizes acid. Subsequent work demonstrated that a large number of mucosal defense mechanisms prevent mucosal damage and maintain mucosal integrity. Vane discovered that the major mechanism by which aspirin and other nonsteroidal antiinflammatory drugs (NSAIDs) produce gastric damage involves inhibition of prostaglandin (PG) synthesis. The concept of cytoprotection developed in the late 1970s and early 1980s by Robert and colleagues sparked tremendous interest in gastric mucosal defense mechanisms. Gastric mucosal injury may occur if noxious factors “overwhelm” an intact mucosal defense or the mucosal defensive mechanisms are impaired.
Gastric Mucosal Defense Mechanisms
Defense mechanisms permit the gastric mucosa to withstand frequent exposure to damaging factors. These include local mechanisms such as the mucus-bicarbonate-phospholipid barrier, continuous cell renewal from mucosal progenitor cells, alkaline tide, and mucosal microcirculation. These concepts have been recently reviewed and illustrated by Laine and colleagues. Fig. 1 depicts the key components of gastric mucosal defense. Various peptides, including gastrin 17, cholecystokinin, thyrotropin-releasing hormone, bombesin, corticotropin-releasing factor (CRF), epidermal growth factor (EGF), peptide YY, neurokinin A analogs, and intragastric peptone, exert gastroprotection, which is abolished by afferent nerve denervation, blockade of calcitonin gene-related peptide receptors, and inhibition of NO synthase.
Ischemia and Reperfusion Injury
Stress-related gastric mucosal injury seems to be related to local ischemia, although progression to significant mucosal injury also requires acid. Critically ill patients developing acute gastric mucosal lesions show a significant decrease in gastric mucosal blood volume compared with controls. Studies in the rat model revealed a significant correlation between blood pressure during hypotension and gastric mucosal blood flow. Mucosal blood flows measured by the hydrogen gas clearance technique in the corpus, antrum, and duodenum of rats all showed a significant linear correlation with mean blood pressure. Blood flow to the mucosa in the stomach and duodenum decreased progressively as systemic blood pressure fell. Significant gastric mucosal lesions occurred only after mean blood pressure and, hence, mucosal blood flow was reduced to less than 40% of baseline values. A rat model of hemorrhagic shock and retransfusion-induced gastric injury revealed that histologic mucosal damage occurred even without acid, although it increased with intragastric instillation of acid in a dose-dependent fashion. In addition, reperfusion after ischemia increased histologic injury compared with ischemia alone, although increasing the duration of ischemia (from 20 to 30 minutes) increased gastric histologic lesions to the same extent as a similar period of retransfusion (10 minutes after 20 minutes ischemia). In the absence of acid, gross gastric lesions were minimal, involving 4% of the body and 3% of the antrum surface area. However, intragastric instillation of acid (0.1 mol/L HCl) markedly increased the gross lesions in a dose-dependent fashion to 53% and 45% of the body and antral surface areas, respectively. These studies imply that splanchnic hypoperfusion in the presence of gastric acid leads to stress-related mucosal damage and suppression of gastric acid would minimize such injuries. This forms the basis for the use of gastric antisecretory agents as prophylactic agents for stress-related mucosal injuries.
Oxidative Stress
Under hypoxic-ischemic conditions, reactive oxygen species (ROS), such as superoxide anions, hydrogen peroxide, and hydroxyl radicals, are rapidly and continuously produced. Oxidative stress resulting from hypoxia and ischemia to the gastric and duodenal mucosa has proved to be an important element in the development and progression of epithelial necrosis and mucosal ulceration. Overproduction of ROS results in oxidative damage, including lipid peroxidation, protein oxidation, and DNA damage, which can lead to cell death. Furthermore, ROS are known to act as second messengers to activate diverse redox-sensitive signaling transduction cascades, including mitogen-activated protein kinases (MAPKs) and downstream transcription factors such as NF-κB and AP-1. These signaling pathways and transcription factors regulate the expression of numerous pro-inflammatory genes and, thereby, lead to the elaboration of chemical and humoral mediators of tissue inflammation and injury. The p38 MAPK is considered important in determining cellular adaptive responses and cell fate. It has been demonstrated that ROS-mediated p38 activation plays an essential role in the pathogenesis of stress-induced gastric inflammatory damage in the rat model of cold immobilization stress. These results suggest that local ROS generation could be an initiating or triggering event in the early phase of stress-induced gastric mucosal injury. The clinically relevant translation of this area of research is the importance of restoring and maintaining systemic and regional circulation and tissue oxygenation in critically ill patients.
The Cyclooxygenase-2 Pathway
Whereas cyclooxygenase-2 (COX-2) has no essential role in the maintenance of gastric mucosal integrity under basal conditions, COX-2 is rapidly induced in a pro-ulcerogenic setting and contributes to mucosal defense by minimizing injury. Ischemia/reperfusion has been shown to increase COX-2 messenger RNA levels in the rat gastric mucosa. It has also been discovered that hypoxia increases expression of the COX-2 gene in human vascular endothelial cells in culture, independent of other stimuli. Therefore, COX-2 expression is assumed to be up-regulated as one of the protective mechanisms when the stomach is exposed to stress, and contributes to mucosal defense by minimizing injury. Therefore, it is reasonable to speculate that COX-2 may prevent gastric mucosal ulcers in mild stress by a phenomenon of adaptive cytoprotection, but may not be able to prevent stress-related mucosal damage in severe stress because of the intensity of the insult and activation of various other pro-ulcerogenic mechanisms. It is understandable why NSAID exposure in the critically ill patient may have profound adverse effects on sustaining the COX-2 defense mechanism.
Endogenous Nitric Oxide and Endothelin-1
Gastric microcirculatory disturbances are involved in the pathogenesis of stress ulcers; however, the vasomodulators causing this vascular instability are not fully understood. Reduced local mucosal nitric oxide (NO; a vasodilator) generation and increased endothelin-1 (a potent vasoconstrictor) levels seem to be involved in events associated with compromise of mucosal blood flow observed in SRMD. In addition, NO plays an important role in the regulation of epithelial cell functions in the GI tract. Gene expression of inducible NO synthase (iNOS) is markedly up-regulated following ischemia/reperfusion and is accompanied by a significant increase in the mucosal NO content. Endogenous NO plays a dual role in ischemia/reperfusion-induced gastric injury; constitutive NO synthase/NO is protective, whereas inducible NO synthase/NO can be pro-ulcerogenic during ischemia/reperfusion. The constitutive NO synthase/NO acts to maintain mucosal integrity through modulating various functions such as mucus secretion, bicarbonate secretion, and mucosal blood flow. In contrast, the inflamed mucosa is associated with recruitment of various inflammatory cells as well as up-regulation of iNOS, cytokines, and adhesion molecules. Under stress conditions, NO produced by iNOS reacts with O 2 − produced by neutrophils to form peroxynitrites. These NO-derived reactive compounds contribute to the nitrosative and oxidative stress burden of vulnerable tissue such as the mucosa of the stomach and duodenum. Thus, the duality of NO function is dependent on the vascular and metabolic integrity of the mucosal and submucosal tissues and the presence or absence of inflammation. Similarly, endothelin-1 levels in plasma and injured mucosa of critically ill patients have been found to be elevated, suggesting endogenous ET-1 may play an important role in the local pathogenesis of stress ulcers.
Mucosal Blood Flow
Critically ill patients may experience a decrease in gastric mucosal blood flow through systemic hypotension and splanchnic hypoperfusion. In rats, gastric lesions began to appear when blood pressure fell to 33% of baseline during hemorrhagic shock. At blood pressure 80% below baseline, 26.8% of the total corpus area developed lesions. Normotensive critically ill septic patients on mechanical ventilation were reported to have a 50% to 60% reduction in gastric mucosal blood flow, measured by reflectance spectrophotometry, compared with controls. Fig. 2 illustrates the consequences of mechanical ventilation in critically ill patients. Splanchnic hypoperfusion in mechanically ventilated patients is caused by decreased cardiac output, release of pro-inflammatory cytokines, and sympathetic system activation. Mechanical ventilation as a risk factor for stress-related mucosal damage is discussed further in next section.
In summary, the etiology and pathophysiology of SRMD is multifactorial and, although several steps in this process have been elucidated, there are additional questions that require further investigation. Ischemia and reperfusion are believed to cause disruption of the mucosal defenses, resulting in mucosal injury and ulceration. The major causative factors of SRMD that have been recognized to date include reduced mucosal blood flow, ischemia, and reperfusion injury. The integrity of the gastric mucosa is maintained by several factors, including the microcirculation and the mucus-bicarbonate gel layer. Maintenance of these functional and structural elements provides nutrient and oxygen delivery to the epithelial cells, neutralization of hydrogen ions to prevent acid back diffusion, and dissipation of reactive substances and other toxic cellular byproducts. Thus, stress-related mucosal damage develops if these elements of protection are either compromised or breached. Tissue PG levels have been shown to be decreased, which in turn impairs mucus replenishment and microcirculation responsiveness. Elevated levels of nitric oxide synthase have also been shown to contribute to reperfusion injury and necrotic cell death, and tissue inflammation response.
Risk factors for stress-induced ulcer bleeding
The frequency of clinically significant stress-related mucosal bleeding in critically ill patients is related to certain risk factors. A prospective multicenter cohort study evaluated potential risk factors for stress ulceration in patients admitted to ICUs. Of 2252 patients, 33 (1.5%) had clinically important bleeding. Two strong independent risk factors for bleeding were identified: respiratory failure (odds ratio [OR] 15.6) and coagulopathy (OR 4.3). Of 847 patients who had one or both of these risk factors, 31 (3.7%) had clinically important bleeding. Of 1405 patients without these risk factors, 2 (0.1%) had clinically important bleeding.
In another multicenter cohort study involving medical and surgical ICUs in six tertiary care Department of Veterans Affairs Medical Centers, patients were evaluated prospectively for the development of acute upper GI hemorrhage while in the ICU. Seventy-six (9%) patients had overt upper GI bleeding and a mortality rate of 49%, compared with a 15% mortality rate in patients who did not bleed ( P <.001). By logistic regression analysis, the following factors were associated with an increased risk of bleeding: acute hepatic failure, prolonged duration of nasogastric tube placement, alcoholism, renal failure, and an increased serum concentration of anti– Helicobacter pylori ( H pylori ) immunoglobulin A. The various risk factors identified from these two studies are listed in Table 1 .
| Risk Factors | Odds Ratio | References |
|---|---|---|
| Respiratory failure | 15.6 | |
| Acute hepatic failure | 6.67 | |
| Coagulopathy | 4.3 | |
| Hypotension | 3.7 | |
| Chronic renal failure | 3.03 | |
| Prolonged duration of NG tube placement | 2.59 | |
| History of alcohol abuse | 2.23 | |
| Sepsis | 2.0 | |
| Helicobacter pylori IgA >1 | 1.92 |
These studies suggest that prophylaxis therapies for bleeding can be safely withheld in critically ill patients who do not have major risk factors. Restricting the use of prophylactic medicinal interventions to patients who have risk factors has not been shown to increase morbidity or mortality in critically ill patients and is more cost-effective.
Related posts:
 The Role of Proton Pump Inhibitors in the Management of Upper Gastrointestinal Bleeding
Management of Massive Peptic Ulcer Bleeding
Predicting Poor Outcome from Acute Upper Gastrointestinal Hemorrhage
The Role of Proton Pump Inhibitors in the Management of Upper Gastrointestinal Bleeding
Management of Massive Peptic Ulcer Bleeding
Predicting Poor Outcome from Acute Upper Gastrointestinal Hemorrhage
 Helicobacter pylori -Negative Nonsteroidal Anti-Inflammatory Drug-Negative Ulcer
Helicobacter pylori -Negative Nonsteroidal Anti-Inflammatory Drug-Negative Ulcer
 Differences in Peptic Ulcer Between the East and the West
Balancing Risks and Benefits of Cyclooxygenase-2 Selective Nonsteroidal Anti-Inflammatory Drugs
Differences in Peptic Ulcer Between the East and the West
Balancing Risks and Benefits of Cyclooxygenase-2 Selective Nonsteroidal Anti-Inflammatory Drugs
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


