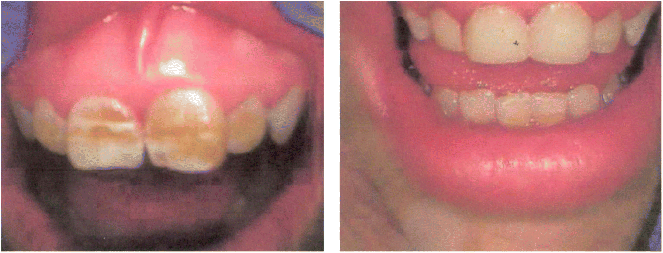
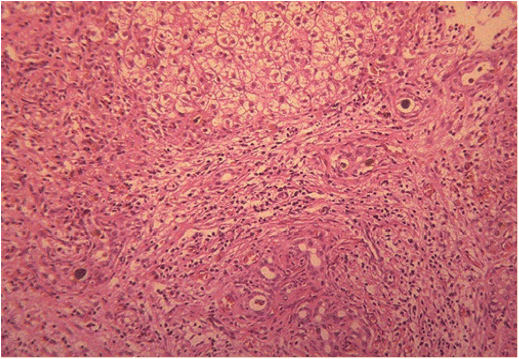
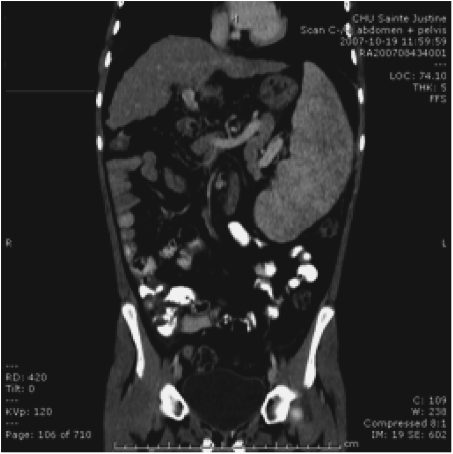
Chapter 28 Alejandro Costaguta1 and Fernando Alvarez2 1Sanatorio de Niños Santa Fe Argentina 2Department of Pediatrics, CHU- Sainte Justine, University of Montreal Quebec Canada The diagnosis and management of cirrhosis in pediatric patients requires special considerations. Different etiologies, unique nutritional requirements, age-specific complications related to different stages of growth and development, as well as size constraints to adapt therapeutic procedures to small children are some of the particularities observed in children. Cirrhosis associated with chronic cholestasis is the most prevalent category at the pediatric age group with the majority of cases resulting from biliary atresia, but cystic fibrosis and progressive familial intrahepatic cholestasis (PFIC) are also seen, among others. Cirrhosis secondary to chronic cholestasis is characterized by a variable time of development, from months to years, and frequently by relatively well-preserved liver synthetic function associated with symptoms and signs of portal hypertension [1]. Children grow and develop due to a complex interplay between hormones and nutrients in which the liver has a central role. Interference in this process often leads to permanent growth and development problems, even after removing the cause (Figure 28.1). Particular concerns have been expressed for some disorders occurring during this critical period of life, which could lead to neurodevelopmental disorders and bone disease [2–4]. Figure 28.1 Teeth in a 12-year-old (left), and a 14-year-old (right) child with biliary atresia transplanted at the age of 6 and 8 years, respectively, because of liver failure. Permanent teeth were stained by deposition of bilirubin in dental hard tissues during their maturation. Small children (weighing 10 kg or less) constitute a significant proportion of pediatric cirrhotic patients. Dealing with specific complications like variceal hemorrhage poses special challenges because of the need to adapt procedures developed for adult patients. Endoscopic variceal ligation or transjugular intrahepatic portosystemic shunt (TIPS) are common examples of therapeutic approaches that are difficult to apply at this age. Management strategies for children with cirrhosis vary according to the natural history of the disease entity. Therefore, this chapter highlights individual disorders leading to cirrhosis in the hope of earlier diagnosis and improved management strategies. This necroinflammatory and obliterative process of the biliary tree, with an incidence of 1 in 10 000–15 000 newborn infants, constitutes the single most common indication for liver transplantation among children. An enigmatic characteristic of this entity is the rapid progression of fibrosis, leading to cirrhosis in the first weeks of life. A combination of genetic and environmental factors has been proposed to explain it, giving rise to a particularly intense fibrogenesis driven by an ongoing immune activation that may persist even after surgical restoration of bile flow [5,6]. After the Kasai portoenterostomy >50% of patients will clear jaundice if surgery is performed before 60 days of age but, unfortunately, almost all will already show an advanced stage of fibrosis or cirrhosis by that time (Figure 28.2) [7]. Figure 28.2 Liver biopsy (H&E staining) from a 2-month-old patient with biliary atresia, showing a rapid evolution to cirrhosis. Practically all patients with biliary atresia develop cirrhosis during the first year of life. Table 28.1 Causes of cirrhosis in children classified according to their pathogenic mechanism. For patients who remain icteric (i.e., total serum bilirubin >2 mg% at 3 months post Kasai), liver transplantation should be offered as soon as possible, because chances of survival beyond the second year are nil [8,9]. There is no other example in hepatology in which the outcome can be predicted with such precision. Children who normalize serum bilirubin levels, on the other hand, face a variable clinical course mainly influenced by the presence of complications from portal hypertension, such as variceal hemorrhage, ascites, and hypersplenism. Bacterial cholangitis is a universal threat, and is associated with the worsening of fibrosis and portal hypertension, especially when recurrent episodes occur. Fifty per cent of this “initial success” group will need liver transplantation in the following years, due to complications from end-stage liver disease leaving only 20–25% of children with biliary atresia to reach adulthood with their native livers, having an almost normal life in the context of compensated cirrhosis [10–12]. This multiorgan disease is produced by mutations in the CFTR gene encoding the membrane protein, CFTR (cystic fibrosis transmembrane regulator) responsible for the efflux of chloride through the cell membrane. As a result, inspissated secretions are produced in exocrine glands with plugging and obstruction followed by activation of the inflammatory cascade, and eventually, secondary infection which stimulates the vicious circle of inflammation and destruction [13]. Liver disease occurs because the CFTR is expressed at the apical level of cholangiocytes, and the consequent biliary obstruction by the inspissated bile is followed by local deposition of collagen, giving rise to the characteristic “focal biliary cirrhosis,” a hallmark of the disease [14]. This initial focal lesion eventually progresses to a multilobular cirrhosis in an estimated 5–10% of all patients with cystic fibrosis. Contributing factors involved in such progression are essentially unknown, but environmental and genetic modifiers have recently been implicated [15,16]. As survival of patients with cystic fibrosis improves due to better control of pulmonary and nutritional complications, liver disease becomes more relevant to the long-term outcome, being the third most common cause of mortality [17,18]. Cystic fibrosis liver disease is difficult to detect in its early stages because of its focal nature, hence an active work-up should be implemented involving physical findings (hepatomegaly, splenomegaly), biochemical tests, and ultrasound examination at regular intervals [19]. Liver biopsy can help in the diagnosis of liver fibrosis, although the patchy distribution of the lesions is frequently misleading and not routinely recommended by all centers caring for these patients. Cohorts followed in this way reveal that around 5% will develop clinically significant liver disease, almost all before the age of 15 which means that severe cystic fibrosis liver disease is a pediatric condition [20]. Ursodeoxycolic acid (UDCA) has been used for many years, in an attempt to halt the progression of the disease. Despite initial enthusiasm produced by some improvement in biochemical liver tests, changes in significant clinical outcomes have not been confirmed in the long term. Starting this drug in earlier stages of disease is an appealing alternative, but better tests are needed to identify patients who could benefit from it. Nevertheless, UDCA is generally used because of its safety profile and theoretical benefits in the absence of better alternatives [14]. Owing to the well-preserved synthetic liver function and multi-organ character of the disease, the best approach to manage complications from portal hypertension in patients with cystic fibrosis is controversial. Variceal hemorrhage represents a life-threatening complication usually controlled by endoscopic and pharmacologic treatments, but its recurrent tendency makes repetitive anesthetic procedures necessary in patients who are poor candidates because of suboptimal pulmonary function [21]. Moreover, hypersplenism tends to be particularly severe in patients with cystic fibrosis, who often exhibit huge and sometimes painful splenomegaly that is not resolved by endoscopic treatment. Surgical shunts or, when feasible, TIPS, offer the possibility of decompressing the portal tree and have been used for a long time in selected patients. However, concerns exist over the possible consequences in the pulmonary vascular bed and the risk of encephalopathy to which patients with cystic fibrosis are especially prone. Liver transplantation has the potential of being “curative” of cystic fibrosis liver disease, but patients should be carefully selected, and only those with a well-preserved pulmonary function should be considered. Results of single liver transplantation are very encouraging, with survival rates similar to other more common indications [17,22]. PFIC constitutes a heterogeneous group of diseases formerly called Byler’s disease and characterized by intracellular cholestasis due to molecular defects interfering with bile formation [23]. The group exhibits signs of cholestasis from the first months of life, evolving rapidly to end-stage liver disease in a significant number of affected children. Biochemical and genetic differences allow classification into three main subtypes with different natural histories, leading to cirrhosis and liver failure mainly in the pediatric age group [24,25]. Affected patients with low gamma-glutamyl transpeptidase (GGT) levels could belong to one of two categories: FIC1 defects (ATP8B1 mutation) or BSEP defects (ABCB11 mutation). In the first group, the disorder is not confined to the liver given the expression of FIC1 in other cell types, particularly enterocytes and pancreatic acinar cells [26]. Phenotypic characteristics include jaundice and pruritus starting in the first months of life, typically with little impairment of liver function for many years, but with a profound impact on the quality of life, similar to recurrent benign cholestasis in the adult, which constitutes the other end of the spectrum [27,28]. Eventually, progressive fibrosis leading to complications ensues, and the decision to proceed to liver transplantation must be carefully considered given the risk of post-transplant complications in this group, like intractable diarrhea, recurrent pancreatitis, and graft steatosis. Partial external biliary diversion has been considered a better approach and should be offered early to these children [29]. Children bearing mutations in BSEP also exhibit normal GGT levels, but their clinical picture tends to be more aggressive, with progressive cholestasis and signs of liver failure during the first years. Therapy with UDCA or external diversion of bile should be offered first to these patients [30,31]. As the disease is confined to the liver, in contrast to FIC1 patients, transplantation is a curative option when decompensation ensues or there is suspicion of hepatocellular carcinoma, which is a worrying complication in the long term [25,32]. Milder forms have also been described [28,33], and adults with mutations in ABCB11 and a picture of benign recurrent cholestasis have been well documented [34], highlighting the many different expressions of this entity. Recurrent cholestasis after liver transplantation has been described, secondary to an immune-related reactivity against the BSEP protein that was not expressed in the native liver [35]. Patients with high GGT levels and a normal biliary tree usually have a defect in the MDR3 gene, giving rise to mutations in the MDR3 transporter which reduces the concentration of phospholipids in bile. Phospholipids are necessary to buffer the detergent effects of bile salts on the cholangiocyte plasma membranes. This explains a picture of generalized damage to the intrahepatic biliary tree, with a characteristic histologic picture of portal fibrosis and cholangiolar proliferation akin to obstructive bile duct diseases [36]. The clinical spectrum of the disease, again, is very broad, from neonatal cholestasis to adult cirrhosis [37]. Affected children exhibit a biochemical picture of cholestasis with variable degrees of jaundice and pruritus in the context of portal hypertension. Management is usually directed to control the complications of portal hypertension and chronic cholestasis [30]. For those who exhibit some degree of liver failure, liver transplantation is an option [38]. As a group, PFIC constitutes a growing category of diseases with a very heterogeneous picture for which a tailored approach should be taken in each case. Genotype–phenotype associations are being unraveled and as more detailed genetic testing is conducted, overlap among different categories is becoming evident, and is the focus of intense research. In addition, studies on protein sorting have led to new forms of therapy [25,39–41]. Progressive cholestasis has been described in children with Langerhans cell histiocytosis who go on to develop primary sclerosing cholangitis (PSC). This uncommon association should be suspected and investigated in every patient having this form of histiocytosis associated with abnormal liver tests (mainly increased GGT levels), as specific treatment protocols can result in stabilization and eventually remission of the liver disease. Progression of the disease leads to cirrhosis and development of life-threatening complications, for which liver transplantation becomes necessary. Survival rates have been comparable to other more common indications, and no relapse of the original disease has been reported so far in the transplanted liver [42]. Although this is typically a pediatric disease, rare cases among adult patients have also been reported [43]. Autoimmune hepatitis (AIH) is a necroinflammatory disease of the liver of unknown etiology progressing, if untreated, to cirrhosis and liver failure in many patients. Characteristic presence of circulating autoantibodies distinguishes two types of AIH: patients with type 1 AIH have anti-smooth muscle antibodies or antinuclear factor, whereas anti-liver-kidney microsome or liver cytosol antibodies define type 2 AIH. Forty percent of type 1 AIH, and 80% of cases of type 2 AIH are diagnosed before the age of 18 years, which means that this is primarily a pediatric disease [44]. AIH is a fluctuating disease characterized by alternating periods of remission and relapses and a silent course occasionally showing a “hepatitis-like” picture with malaise, jaundice, and dark urine. Affected patients commonly develop a firm liver, with a prominent left lobe. Splenomegaly, spider nevi, palmar erythema, and occasionally ascites are signs of chronicity and should alert the physician to consider AIH among differential diagnoses. Acute liver failure, or acute decompensation of a chronic condition, can also be a form of presentation, in approximately 10% of patients [45]. Unfortunately, in a substantial number of patients, the interval from the first symptoms to the diagnosis is longer than expected, allowing the inflammatory liver injury to progress to more advanced stages of fibrosis before an effective treatment is implemented (Figure 28.3) [46]. Figure 28.3 Magnetic resonance image of the abdomen of a 14-year-old girl with late diagnosis of autoimmune hepatitis showing nodular liver atrophy and large splenomegaly secondary to portal hypertension (spleen length 173 mm).
Special Considerations in Children
Introduction
Particular Liver Diseases in Children (Table 28.1)
Chronic Cholestasis
Biliary Atresia
Cholestatic causes
Biliary atresia
Cystic fibrosis
Progressive intrahepatic familial cholestasis
Histiocytosis X
Liver inflammatory diseases
Hepatitis
Metabolic disorders
Alpha-1-antitrypsine deficiency
Wilson’s disease
Glycogenosis types III and IX
Glycogenosis type IV
Tyrosinemia
Niemann–Pick type C
Toxic disorders (nutritional)
Total parenteral nutrition (TPN)
Cystic Fibrosis
Progressive Familial Intrahepatic Cholestasis
Langerhans Cell Histiocytosis
Chronic Liver Inflammation
Autoimmune Hepatitis
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


