Renal Acid-Base Transport
Thomas D. DuBose Jr.
Amanda K. Goode
ACID-BASE HOMEOSTASIS
The body maintains systemic acid-base homeostasis (a pH in the range of 7.35 to 7.45) in two ways: (1) through chemical buffering in extracellular fluid (ECF) and intracellular fluid (ICF), and (2) through physiologic regulation controlled by the metabolic, renal, and respiratory systems. The central nervous and respiratory systems control CO2 tension (PCO2), and the kidneys regulate the plasma HCO3– concentration. Buffers in the ECF and ICF guard against acid and base retention. These processes serve to dispose of carbonic and nonvolatile acids on a daily basis and pathologic quantities of acid and alkali as needed. This chapter briefly reviews the role of chemical buffers, but focuses primarily on the renal transport and regulatory processes that maintain acidbase balance.
Buffer Systems
The body’s buffer systems keep the blood from becoming too basic or too acidic by combining with or releasing H+, and thus are comprised of a base (i.e., an H+ acceptor) and an acid (i.e., an H+ donor). The most prodigious base is bicarbonate (HCO3-) and the most common acid is carbonic acid (H2CO3). Because buffer systems attenuate system changes, acid or base loads in the presence of a buffer cause smaller changes in the pH than if the buffer were absent. During homeostasis, the extracellular H+ concentration ([H+]e) is constant. H+ concentration can be expressed either directly as [H+] or indirectly as pH.
When CO2 is dissolved in water, H2CO3 is formed according to the reaction
The rate of this reaction, in the absence of the enzyme carbonic anhydrase, is slow, with a half-life of about 8 seconds at 37°C. The importance of carbonic anhydrase in bicarbonate equilibrium in the kidneys and lungs are discussed in the following paragraphs. The major portion of CO2 remains as dissolved CO2; only about 1 part in 1,000 forms H2CO3, a nonvolatile acid. Because H2CO3 is a weak acid, it dissociates rapidly to yield H+ and HCO3-.
Because the concentration of H2CO3 remains low and proportional to the concentration of dissolved CO2, Eqs 1 and 2 can be combined and treated as a single reaction:
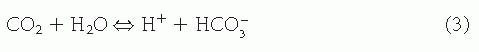
The equilibrium constant for this reaction is given by

Defining K’ = K[H20] as the apparent equilibrium constant and using Eq. 4,

Taking logarithms of both sides of Eq. 5 and recognizing that pK’ = logl0(K’), the familiar Henderson-Hasselbalch equation is derived:
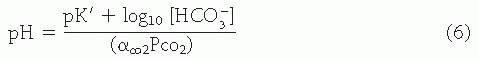
Using pK’ = 6.1 in Eq. 6, the Henderson equation is derived, which may be used in the clinical interpretation of acid-base data:
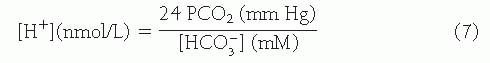
Physiologic Processes that Protect the Plasma Bicarbonate
Three physiologic processes mitigate changes in the HCO3-/ CO2 ratio: (1) metabolic regulation, (2) respiratory regulation, and (3) renal regulation. Metabolic regulation is
of minor importance in terms of the overall physiologic regulation of acid-base balance. Some enzymes are regulated by changes in blood pH. For example, the activity of phosphofructokinase, the pivotal enzyme in the glycolytic pathway, is inhibited by low pH and enhanced by high pH.
of minor importance in terms of the overall physiologic regulation of acid-base balance. Some enzymes are regulated by changes in blood pH. For example, the activity of phosphofructokinase, the pivotal enzyme in the glycolytic pathway, is inhibited by low pH and enhanced by high pH.
Because, under most circumstances, respiratory CO2 excretion and CO2 production are matched, the usual steady state arterial PCO2 (PaCO2) is maintained at 40 mm Hg. Underexcretion of CO2 produces hypercapnia, and overexcretion produces hypocapnia. Production and excretion are again matched but at a new steady state PCO2. Therefore, PaCO2 is regulated primarily by neurorespiratory factors and is not subject to regulation by the rate of metabolic CO2 production. Hypercapnia is primarily the result of hypoventilation, not increased CO2 production. Increases or decreases in PCO2 represent derangements of control of neurorespiratory regulation or can result from compensatory changes in response to a primary alteration in the plasma HCO3– concentration.
Sources of Endogenous Acids
Pathologically, acid loads may be derived from endogenous acid production (e.g., generation of ketoacids and lactic acids) or loss of base (e.g., through diarrhea) or from exogenous sources (e.g., ammonium chloride or toxin ingestion). Under normal physiologic circumstances, a daily input of acid derived from the diet and metabolism confronts the body. The net result of these processes amounts to about 1 mEq of new H+ per kilogram per day entering the ECE1,2,3,4
Sulfuric acid is formed when organic sulfur from methionine and cysteine residues of proteins are oxidized to SO42-. The metabolism of sulfur-containing amino acids is the primary source of acid in the usual Western diet, accounting for approximately 50%. The quantity of sulfuric acid generated is equal to the SO24– excreted in the urine.
Organic acids are derived from intermediary metabolites formed by the partial combustion of dietary carbohydrates, fats, and proteins as well as from nucleic acids (uric acid). Organic acid generation contributes to net endogenous acid production when the conjugate bases are excreted in the urine as organic anions. If full oxidation of these acids can occur, however, H+ is reclaimed and is eliminated as CO2 and water. The net amount of H+ added to the body from this source can be estimated by the amount of organic anions excreted in the urine.
Phosphoric acid can be derived from hydrolysis of PO34 esters in proteins and nucleic acids if it is not neutralized by mineral cations (e.g., Na+, K+, Mg 2+). The contribution of dietary phosphate to acid production is dependent on the kind of protein ingested. Some proteins generate phosphoric acid, whereas others generate only neutral phosphate salts. 1,2,3,4 Hydrochloric acid is generated by the metabolism of cationic amino acids (lysine, arginine, and some histidine residues) into neutral products. Other potential acid or base sources in the diet can be estimated from the amount of unidentified cations and anions ingested.
Potential sources of bases are also found in the diet (e.g., acetate, lactate, citrate) and can be absorbed to neutralize the acid loads from the three sources just mentioned. The net base absorbed by the gastrointestinal tract is derived from the anion gap (AG) of the diet minus that of the stool. Acid production is partially offset by HCO3-, which is produced when organic anions combine with H+ and are oxidized to CO2 and H20 or when dibasic phosphoesters combine with H+ during hydrolysis. The gastrointestinal tract may modify the amount of these potential bases reabsorbed under particular circumstances of acidosis or growth. It has been confirmed in patients ingesting an artificial diet that urinary net acid excretion is equal to urinary [(SO24-) + organic A- + dietary phosphoester-derived H+].1,2,5 Therefore, in summary, the metabolism of certain proteins, nucleic acids, and small fractions of lipids and certain carbohydrates generate specific organic acids that cannot be burned to C02 (e.g., uric, oxalic, glucuronic, hippuric acids). In addition, the inorganic acids H2SO4 and H3P04, derived respectively from sulfur-containing amino acids and organophosphates, are excreted by the kidneys or the gastrointestinal tract.
Impact of Daily Metabolism on Acid-Base Balance
Human subjects ingesting a typical Western diet are confronted, under most physiologic circumstances, with an acid challenge. Metabolism generates a daily load of relatively strong acids (lactate, citrate, acetate, and pyruvate), but under physiologic circumstances, in the steady state, metabolic production and consumption are matched. However, if production and consumption rates become mismatched, organic acids can accumulate (e.g., lactic acid accumulation with anoxia or ischemia). These acids are buffered by HCO3– in the ECF, causing a decline in plasma HCO3– concentration as the organic acid concentration increases. During recovery, these organic acids reenter the metabolic pathways to CO2 production, the removal of H+, and the generation of HCO3-unless the organic anions are excreted (e.g., ketonuria), and thereby are no longer available for regeneration of HCO3–.
Both metabolic and renal regulatory mechanisms protect a normal HCO3– concentration in blood (25 mEq per liter) despite the daily addition of acid (or alkali) to the ECE Although the buffering capacity of the body is magnified severalfold by respiratory adjustments in PaC02, primary changes in PaC02 may result in acidosis or alkalosis, depending on whether CO2 is elevated above or depressed below the normal value: 40 mm Hg (respiratory acidosis and respiratory alkalosis), respectively. A primary change in the plasma HCO3– concentration as a result of metabolic production or the retention or excretion of an acid or base by the kidney evokes a ventilator response because a decrease or increase in pH is sensed by chemoreceptors in the circulation and signals the respiratory center to increase or decrease minute ventilation. The respiratory response to acidemia (increase ventilation
and decrease PCO2) or alkalemia (decrease in ventilation and increase in PCO2) thereby blunts the change in blood pH that would occur otherwise. Such respiratory alterations that adjust blood pH toward normal are referred to as secondary or compensatory alterations, because they occur in response to primary metabolic changes. While helpful, respiratory compensation to metabolic acidosis or alkalosis is never sufficient to return blood pH to normal. Therefore, the kidneys must play a very significant role in adjustments to metabolic disturbances by altering bicarbonate reabsorption and net acid excretion.
and decrease PCO2) or alkalemia (decrease in ventilation and increase in PCO2) thereby blunts the change in blood pH that would occur otherwise. Such respiratory alterations that adjust blood pH toward normal are referred to as secondary or compensatory alterations, because they occur in response to primary metabolic changes. While helpful, respiratory compensation to metabolic acidosis or alkalosis is never sufficient to return blood pH to normal. Therefore, the kidneys must play a very significant role in adjustments to metabolic disturbances by altering bicarbonate reabsorption and net acid excretion.
RENAL PARTICIPATION IN ACID-BASE HOMEOSTASIS
Although temporary relief from changes in the pH of body fluids may be derived from chemical buffering or respiratory compensation, the ultimate defense against the addition of nonvolatile acid or of alkali resides in the kidneys. The addition of a strong acid (e.g., HA) to the ECF titrates plasma HCO˜3-:
The CO2 is expired by the lungs, and body HCO3-declines. This process occurs constantly as endogenous metabolic acids are generated. To maintain a normal plasma HC 03- in the face of the constant accession of metabolic acids, the kidneys must (1) conserve the HCO3– present in the filtered load (through reclamation), and (2) regenerate the HC 03- decomposed by reaction with metabolic acids (Eq. 8).
The first function of the kidneys in maintaining acidbase homeostasis is to reclaim or reabsorb filtered HCO3-. The kidneys filter approximately 4,000 mEq of bicarbonate daily (defined as the product of the glomerular filtration rate [GFR] and the plasma HCO3– concentration), and the excretion of any significant portion of this leads to metabolic acidosis. The renal tubules essentially reabsorb all of the filtered HCO3-, or may excrete excessive HCO3– when needed, to maintain the normal plasma HCO3– concentration of 25 mEq per liter.
HCO3– reclamation is accomplished principally in the proximal tubule. There is a subsequent contribution by the loop of Henle and a minor contribution by more distal nephron segments. Under most circumstances, the filtered load of HCO3– is absorbed almost completely, especially during an acid load. Nevertheless, when less acid is generated or if the plasma HCO3– concentration increases above the normal value of 25 mEq per liter, HCO3– will be excreted efficiently into the urine.
The generation of acid in the body by metabolism is referred to as net acid production. Because a typical Western diet generates fixed acids at 50 to 70 mEq per day, net acid excretion must be affected to maintain acid-base balance. Therefore, net acid excretion approximates 50 to 70 mEq per day. If acid production remained stable and unabated by net acid excretion, metabolic acidosis would ensue. Conversely, an increase in net acid excretion above the level of net acid production results in metabolic alkalosis. Each milliequivalent of net acid excreted corresponds to 1 mEq of HCO3– returned to the ECE This process is called HCO3-regeneration and is necessary for replacing the HCO3– lost by the entry of fixed acids into the ECF or, less commonly, replacing the HCO3– excreted in stool or urine.
To prevent metabolic acidosis, this excreted acid must be recovered. The kidney recovers this acid through the second process, HCO3– regeneration, which is represented by the renal output of acid or net acid excretion (NAE), or the sum of ammonia (NH3)/ammonium (NH+4) plus excreted titratable acids (TA) minus any bicarbonate excreted.
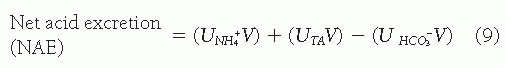
where V is urinary volume per 24 hours and U refers to the concentration of the moiety in the urine.
TA excretion in the urine is represented by buffers that are filtered at the glomerulus and then titrated to acid moieties in the tubule lumen. Because renal H+-secretory mechanisms cannot generate such steep pH gradients between the cell (pH ˜-7.3) and lumen, excretion of 70 mEq of acid per day requires that most of this acid be buffered in the luminal fluid.
Buffers with a pK between 5.0 and 7.4, the pH range of the luminal fluid, are most effective chemically. In this regard, the most abundant and effective buffer in the urine is the phosphate buffer pair, H2PO4–—HPO24-, with a pK of 6.9. The excretion of phosphate, the major titratable buffer, is usually regulated in accordance with phosphate homeostasis. Phosphate excretion increases modestly in metabolic acidosis, but the magnitude of the increase is limited by the filtered load of phosphate and, therefore, is significantly regulatable. In general, the buffers responsible are the NH3—-NH+4 and titratable buffer, such as HPO24-—H2PO–4, systems. Physiologically, the most significant of these is the NH3-NH+4 system.
Ammoniagenesis
Ammoniagenesis by protein catabolism is balanced by the generation of new HCO3– through renal NH4+ and TA excretion (Fig. 7.1A). The products of these neutral reactions are HCO3– and NH+4. Most ammoniagenesis occurs in the proximal tubule. NH+4 produced by the metabolism of amino acids reacts with HCO3– or forms urea and thus has no impact on acid-base balance. A portion of this NH4+ is diverted to glutamine synthesis, the amount of which is regulated by pH. Acidemia promotes and alkalemia inhibits glutamine synthesis. Each glutamine molecule metabolized produces two NH+4 and two HCO3– molecules (Fig. 7.1A) (water, CO2, and glucose are also formed). This breakdown occurs as follows: glutamine is transported into the proximal
tubule across the apical and basolateral membranes and is then transported into the mitochondria. In the mitochondria, an NH+4 molecule is formed when glutamine is deaminated by glutaminase to glutamate. When glutamate is then deaminated to a-ketoglutarate, a second NH+4 molecule is formed. Finally, the a-ketoglutarate molecule is metabolized in the Krebs cycle to CO2, H20, glucose, and the two HCO3-molecules. Glutamine deamination in the kidney is highly regulated by systemic pH, so that acidemia augments and alkalemia inhibits NH4+ and HCO3– production. Hepatic regulation of NH4+ metabolic pathways appears to facilitate glutamine production when NH+4 excretion is stimulated by acidemia or, conversely, blunts glutamine production when excretion is inhibited by alkalemia.5
tubule across the apical and basolateral membranes and is then transported into the mitochondria. In the mitochondria, an NH+4 molecule is formed when glutamine is deaminated by glutaminase to glutamate. When glutamate is then deaminated to a-ketoglutarate, a second NH+4 molecule is formed. Finally, the a-ketoglutarate molecule is metabolized in the Krebs cycle to CO2, H20, glucose, and the two HCO3-molecules. Glutamine deamination in the kidney is highly regulated by systemic pH, so that acidemia augments and alkalemia inhibits NH4+ and HCO3– production. Hepatic regulation of NH4+ metabolic pathways appears to facilitate glutamine production when NH+4 excretion is stimulated by acidemia or, conversely, blunts glutamine production when excretion is inhibited by alkalemia.5
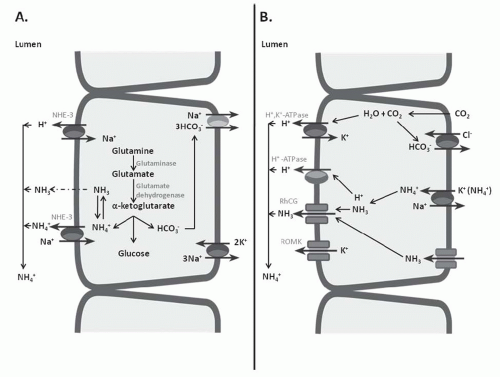 FIGURE 7.1 Cell models of ammonia synthesis and excretion pathways. A: A proximal convoluted tubule. Ammonia is derived from glutamine precursors to produce 2 NH4+ and 2 HCO3– molecules through an enzymatic pathway activated by acidemia and hypokalemia and inhibited by alkalemia and hyperkalemia. B: A type A intercalated cell in the collecting tubule. Ammonium enters across the basolateral membrane through the substitution of K+ for NH+4 in K+ conductance and is secreted across the apical membrane via renal outer medullary potassium (ROMK) or RhCG (see text). In both A and B, NH3 diffusion coupled with H+ secretion traps NH+4 in the tubule lumen. |
Renal Excretion of Ammonia/Ammonium
The ultimate control, however, resides in the renal excretion of NH+4, because the NH+4 must be excreted to escape entry into the hepatic urea synthetic pool. Hepatic urea synthesis would negate the new HCO3– realized from a-ketoglutarate in the kidney. Thus, ammoniagenesis does not contribute to acid-base homeostasis until the urine is acidified and the HCO3– produced by glutamine metabolism is added to the ECE In order for this to occur, the ammonia synthesized in the proximal tubule has to be transported to the terminal nephron segments and is then excreted by the kidney (the pathway is described in detail in the following paragraphs). This ammonia transport occurs through both nonionic diffusion (for NH3) and ionic transport (for NH+4). NH3 quickly and easily diffuses across the plasma membrane to compartments of lower pH, where it is rapidly converted to NH4+, thus leading to accumulated NH+4 in acidic compartments. NH+4 can diffuse across tight junctions or can substitute on transporters such as the apical membrane Na+-H+ (NHE-3) antiporter (as Na+-NH4+ exchange).6 In general, NH3 has a higher membrane permeability than NH+4, but the concentration of NH+4 exceeds that of NH3 by approximately 100-fold at a pH 7.4 (pK 9.3). Thus, the relative
permeability of the two moieties defines whether either crosses membranes by nonionic NH3 diffusion or ionic N H4+ transport; for example, if the NH+4 moiety is transported in a specific nephron segment as in the thick ascending limb of the loop of Henle by substitution for potassium on the Na+ -K+ -2C1- cotransporter.
permeability of the two moieties defines whether either crosses membranes by nonionic NH3 diffusion or ionic N H4+ transport; for example, if the NH+4 moiety is transported in a specific nephron segment as in the thick ascending limb of the loop of Henle by substitution for potassium on the Na+ -K+ -2C1- cotransporter.
The first step of ammonia transport and urinary excretion following ammoniagenesis in the proximal tubule is for the ammonia to enter the proximal tubule lumen, and it can do so in two ways. Because the luminal pH is lower than cell pH, much of the NH3 is diffused into the tubule fluid and is converted into NH4+. The apical membrane Na+ -H+ antiporter also plays an important role in NH3-NH+4 transport (Fig. 7.1A).7,8 Transport mechanisms for NH3-NH+4 in more distal segments function to ensure that the NH3– NH4+ issuing out of the proximal tubule fluid is excreted into the urine in a regulated fashion.
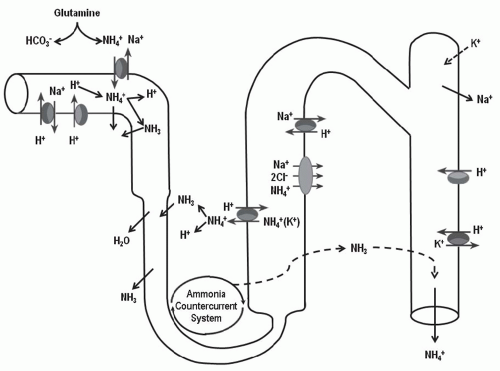 FIGURE 7.2 Nephron sites of ammonia production, secretion, transport, and ultimately, excretion. Note that approximately 75% of ammonia/ammonium delivered to the loop of Henle is transported into the medullary interstitium and is accumulated through countercurrent multiplication, thus resulting in higher concentrations from the cortex to the medulla. In acidosis, both the production of ammonia/ammonium and the transport, multiplication, and excretion of NH+4 is augmented. Therefore, the physiologic response of the kidney to an acid load is to increase bicarbonate reabsorption and ammonia production and excretion. The resulting urine should demonstrate a pH <5.5 and a higher concentration of ammonium. If these conditions are met, a normal gap metabolic acidosis would be nonrenal in origin. |
From the lumen, ammonia enters the medullary interstitium (Fig. 7.2) via the thick ascending limb of the loop of Henle. If ammonia did not enter the interstitium, it would return to the distal nephron and diffuse back into the blood, thereby failing to be excreted in the urine. The thick ascending limb of the loop of Henle also creates a “corticomedullary concentration gradient” for NH3-NH+4 The NH3-NH+4 transported into the medullary interstitium accumulates at a higher concentration by countercurrent multiplication and can reenter the collecting tubule for a final excretion in a more regulated way. Nonionic diffusion of NH3 into the medullary interstitium is not possible because the thick ascending limb’s apical membrane is highly impermeable to NH3.9 NaCl transport enables the ionic transport of NH+4 into the interstitium. NH+4 can substitute for K+ on the Na+-K+-2C1- cotransporter, or it can be transported via the renal outer medullary potassium (ROMK) channel for transport across the apical membrane (Fig. 7.1B). Alternatively, lumen-positive voltage can drive NH+4 diffusion across the tight junction. NH+4 that enters the thick ascending limb cell can exit the basolateral membrane by nonionic diffusion driven by cell-to-interstitial NH3 concentration gradient. Once in the interstitium, the NH3
will again accept a H+, forming NH+4. Furosemide, which inhibits the Na+-K+-2C1- cotransporter and secondarily inhibits the lumen-positive voltage, significantly inhibits all three mechanisms of thick ascending limb NH3-NH+4 absorption. 6,10,11
will again accept a H+, forming NH+4. Furosemide, which inhibits the Na+-K+-2C1- cotransporter and secondarily inhibits the lumen-positive voltage, significantly inhibits all three mechanisms of thick ascending limb NH3-NH+4 absorption. 6,10,11
Through these processes, NH3-NH+4 accumulates in the renal medulla (Fig. 7.2). The countercurrent system concentrates the solute toward the tip of the medulla and preserves the cortical-to-medullary gradient. Blood in the descending vasa recta has lower NH3-NH+4 concentrations than the surrounding interstitium, allowing net entry. Conversely, blood in the ascending vasa recta has a higher NH3-NH+4 concentration than the surrounding interstitium, causing net efflux. In patients with an intact urinary concentrating system and medullary architecture, the net result is a steep gradient, with the highest concentrations of NH3-NH+4 in the inner medullary interstitium.
Ultimately, NH3-NH+4 is transported from the medullary interstitium into the lumen of the collecting tubule for final urinary excretion (Figs. 7.1B and 7.2). The apical and basolateral membranes of the medullary collecting tubule are permeable to NH3, allowing nonionic diffusion to be driven by the low pH of the collecting tubule fluid. In addition, NH+4 can be transported from the medullary interstitium into the collecting tubule cell on the Na+, K+-ATPase. 12 Once inside the cell, the dissociation of NH+4 into NH3 and H+ provides a source of H+ for transport into the luminal fluid by the vacuolar H+-ATPase. H+ secreted into the luminal fluid will combine with secreted NH3 to form NH+4, maintaining the gradient for nonionic NH3 diffusion into the luminal compartment, which has a much lower pH, and this way NH+4 is “trapped” in tubule fluid and is swept into the collecting system. Medullary interstitial NH3-NH+4 concentrations are determined by the rate of ammonia synthesis in the proximal tubule, the efficiency of ammonia transport into the lumen of the proximal tubule and subsequently into the medullary interstitium in the thick ascending limb, and the efficiency of countercurrent trapping of ammonia in the medullary interstitium. Both ammonia synthesis and each step in the highly integrated response by the nephron to excrete ammonia are upregulated by metabolic acidosis and chronic hypokalemia, and are inhibited by alkalosis and hyperkalemia (see Chapter 18).
The Rh glycoprotein (RhCG/Rhcg), which is expressed not only in both principal and intercalated cells but also in the distal convoluted tubule, connecting tubule, initial collecting tubule, cortical collecting duct, and the outer and inner collecting ducts, is now thought to be critical for the excretion of ammonia by the kidney (Fig. 7.1B).13,14 RhCG expression is greater in A-type intercalated cells than in principal cells but is not detectable in B-type intercalated cells.15 RhCG is thought to play a role in renal ammonia excretion because of some recent genetic studies. Biver and colleagues16 found that global RhCG deletion impaired ammonia excretion in the urine and decreased basal ammonia excretion in response to metabolic acidosis. Similar findings were observed when RhCG was deleted only from the collect ducts.17 In summary, that both intercalated and principal cells express RhCG and that metabolic acidosis increases RhCG in both cell types suggests that RhCG contributes to transepithelial ammonia secretion.16
Therefore, the sum of TA and ammonia/ammonium excretion is represented, as noted previously, as net acid excretion (Eq. 9). All three components of net acid excretion are dependent on the operation of an H+ secretory mechanism into the tubule fluid. Therefore, increases in the rate of H+ secretion will increase urinary ammonium and TA excretion and will lower urinary HCO3– excretion. A protein-rich diet leads to acid production, so the kidneys must both reclaim the filtered bicarbonate and also excrete an acid equivalent to that produced in the diet. In this way, in a steady state, net acid production equals net acid excretion, and acid-base balance is maintained. In the presence of nonprotein-rich diets, the tubules do not reclaim all of the filtered bicarbonate and they decrease production of new bicarbonate in order to maintain acid-base homeostasis.
SPECIFIC MECHANISMS OF H+-HCO3-TRANSPORT ALONG THE NEPHRON
How the transport of H+ and HCO3– in the nephron occurs is generally defined by the specific characteristics of each epithelium and cell type within the nephron segments. Bicarbonate reabsorption is largely the responsibility of the proximal tubule and net acid secretion is largely of the distal tubule. A number of transport mechanisms across either the apical (for H+) or basolateral (for alkali) membranes have been identified and are detailed in the following paragraphs.
Apical Membrane H+-Transport Mechanisms
Renal tubule cells use both primary and secondary transport mechanisms for active H+ secretion. A primary transport couples the metabolism to a transport mechanism, whereas a secondary transport couples the transporter to the metabolism, which generates a concentration gradient for a solute.
There are two mechanisms for primary active H+ secretion, both of which are directly related to the metabolism of adenosine triphosphate (ATP) to adenosine diphosphate (ADP) and inorganic phosphate (Pi). The first and quantitatively most important mechanism involves the vacuolar H+-ATPase, or V-type ATPase, a multisubunit protein complex that secretes H+, 18 and is electrogenic, thereby generating a positive charge in the tubule lumen. The ability of the H+-ATPase to affect H+ secretion depends on where it is located; when it is in the proximal tubule, it minimally mediates H+ secretion into the lumen, but in type A (acid secreting) intercalated cells of the collecting tubule, it significantly mediates H+ secretion into the lumen. This
mechanism is able to adapt as needed by increasing mRNA and protein abundance to protect against metabolic and respiratory acidosis.
mechanism is able to adapt as needed by increasing mRNA and protein abundance to protect against metabolic and respiratory acidosis.
The second mechanism for primary active H+ secretion involves P-type ATPases. The P-type ATPases, which are isoforms of the H+ -K+ -ATPase (HKα1 , the gastric isoform, and HKα2, the colonic isoform), participate importantly in H+ transport by the collecting tubule. These P-type ATPases are coupled to ATP metabolism, have a phosphorylated high-energy intermediate, and possess α and β subunits. Both the gastric and colonic isoforms are expressed on the apical membranes of type A intercalated cells (IC) in the cortical collecting tubule, and the outer and inner medullary collecting tubules.19,20,21,22,23,24 These transport processes are electroneutral, mediating H+ secretion across the apical membrane into the tubule lumen in exchange for K+ entry (into the cell). Both isoforms react to metabolic or respiratory acidosis by increasing in abundance and activity. Alternatively, the two types of H+ -transporters are also located on the basolateral membranes of type B (bicarbonate secreting) ICs in the cortical and outer collecting tubule.25,26,27,28,29 The H+ -ATPase or H+, K+ -ATPase in this location mediates H+ transport across the basolateral membrane into the interstitium.
The secondary active H+ transporter is an isoform of the Na+-H+ antiporter, NHE-3. Although there are eight known isoforms of the Na+ -H+ antiporter, NHE-3 (SLC9A3) is the most important for bicarbonate reabsorption. It is located in the S1 and S2 proximal tubule and thick ascending limb of the loop of Henle on the apical membrane and mediates H+ secretion into the tubule fluid.30,31,32,33
Basolateral Bicarbonate Transport
Two transporters mediate passive base efflux along the nephron. One transporter is the electroneutral Cl-/HCO3-exchanger, which carries HCO3– out and C1- into the cell across the basolateral membrane in the S3 segment of the proximal tubule, the thick ascending limb of the loop of Henle, and the cortical and medullary collecting tubules. This Cl-/HCO3– exchanger is a member of the anion exchanger 1 (AE1) family and is a truncated form of the red cell AE1 Cl-/HCO3– exchanger,34,35,36,37 or kAE1 (SLC4A1). The mRNA lacks the first three exons of red cell mRNA,38 but the net result of this truncation is a protein with similar transport characteristics but altered cytoskeletal interactions. C1- enters the cell in exchange for HCO3– and exits the basolateral membrane through a C1- conductance.39,40
The second is the sodium bicarbonate cotransporter (NBC), of which there are four isoforms. The NBCel (SLC4A4) isoform is the most important for base efflux, mediating the majority of base efflux (˜-80%) out of the cell and into the interstitium.41,42,43,44 It is present on the basolateral membrane of the proximal tubule and is electrogenic because it transports three base molecules with one Na+ ion (Na+/ n HCO3- or SLC4A4).41,42,43,44
SEGMENTAL CONTRIBUTION TO BICARBONATE ABSORPTION AND ACIDIFICATION OF THE URINE Proximal Tubule
The proximal tubule reabsorbs approximately 80% of the filtered HCO3– load; the HCO3– concentration at the end of the proximal tubule is approximately 8 mEq per liter and the pH is 6.7.45,46




Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
