Chapter 41 Primary sclerosing cholangitis
Overview
Primary sclerosing cholangitis (PSC) is a chronic, cholestatic liver disease of unknown etiology characterized by diffuse inflammatory destruction of intrahepatic and/or extrahepatic bile ducts that results in bile stasis, hepatic fibrosis, cirrhosis, and end-stage liver disease. Despite the increased awareness of PSC, the exact etiology and an effective medical therapy have yet to be identified. The disease is associated with specific human leukocyte antigen (HLA) haplotypes and has a close association with inflammatory bowel disease (IBD), which suggests that PSC is an immune-mediated disorder; but unlike other typical autoimmune diseases, there is a 2:1 male predominance. Life expectancy is reduced in PSC patients, because disease progression often results in complications of liver failure and cholangiocarcinoma (see Chapters 50A, 50B, and 50C). Among eligible candidates, surgical therapy has moved largely to orthotopic liver transplantation (OLT), as it remains the only effective, life-extending treatment for advanced-stage PSC (see Chapter 97A).
Epidemiology and Demographics
PSC mainly affects males during the fourth decade of life (Angulo & Lindor, 1999; Chapman, 2003). In the United States, population-based studies reported an age-adjusted incidence for PSC of 1.25 per 100,000 men and 0.54 per 100,000 women per year (Bambha et al, 2003). In the same study, the calculated prevalence of PSC was 20.9 and 6.3 per 100,000 men and women, respectively (Bambha et al, 2003). The worldwide prevalence and geographic distribution of PSC are undefined, as most epidemiologic data come from Northern Europe and the upper Midwest in the United States. Of interest is the strong association of PSC with IBD. Approximately 75% to 80% of Northern European patients with PSC suffer from IBD, with chronic ulcerative colitis (CUC) being most common, seen in about 90% (Chapman et al, 1980; Olsson et al, 1991; Wiesner et al, 1989). Conversely, only approximately 5.5% of patients with CUC have PSC (Olsson et al, 1991).
Smoking is the only environmental factor known to influence PSC susceptibility, and it is associated with a reduced risk of PSC. In a study from The Netherlands, the incidence of current smoking was 19% in patients with PSC compared with 38% in control subjects (Van Erpecum et al, 1996). Similarly, a U.S. study showed that 4.9% of patients with PSC were reported to smoke compared with 26.1% of a control population matched for age and gender. This reported difference was not attributable to the prevention of CUC (Loftus et al, 1996b). Nevertheless, smoking and alcohol consumption should be discouraged in PSC patients, because both are associated with the development of cholangiocarcinoma in this population (Bergquist et al, 1998; Chalasani et al, 2000).
Adenocarcinoma of the bile ducts appears in 8% to 15% of patients with PSC, rendering it a premalignant condition (Chalasani et al, 2000; De Groen et al, 1999; Foutch et al, 1990; see Chapters 50A, 50B, and 50C). In a recent study, 161 PSC patients without evidence of cholangiocarcinoma upon entry were followed over a median of 11.5 years. During that period, 59 patients (36.6%) died, 50 patients (31.1%) underwent liver transplantation, and 11 patients (6.8%) had cholangiocarcinoma develop (Burak et al, 2004). No association was found between the duration of PSC and the incidence of cholangiocarcinoma, and multivariate analysis revealed that variceal bleeding was the only significant risk factor for the diagnosis of cancer (risk ratio, 24.2; 95% confidence interval, 3.3% to 67%) (Burak et al, 2004).
Clinical Presentation
PSC can affect any age group, but it is more common among men in their fourth decade of life (Wiesner & LaRusso, 1980; MacCarty et al, 1983). PSC can also occur in childhood, and children with PSC frequently have liver disease with features similar to autoimmune hepatitis (AIH) (El-Shabrawi et al, 1987; Wilschanski et al, 1995). In fact, the overlap between PSC and AIH in children can be as high as 35% (Feldstein et al, 2003). In contrast, overlap of PSC and AIH affects only about 5% of adults (Kaya et al, 2000; Van Buuren et al, 2000).
The clinical presentation of PSC varies depending on the disease stage at the time of diagnosis. PSC can be diagnosed in asymptomatic individuals who come to medical attention because of abnormal liver function tests. Approximately 15% to 40% of patients are asymptomatic at the time of diagnosis (Talwalkar & Lindor, 2005). There are no pathognomonic signs or symptoms when the disease becomes clinically apparent. The symptomatic patient may be seen initially with signs, symptoms, and complications of cholestatic liver disease or hepatic failure. In a recent study, the most common first symptoms were abdominal pain (20%), pruritus (10%), diarrhea (8%), jaundice (6%), fatigue (6%), and fever (4%) (Kaplan et al, 2007). Symptoms of bacterial cholangitis are uncommon, unless dominant biliary strictures or biliary stones are present.
Physical examination may reveal jaundice, hepatomegaly, splenomegaly, and excoriations. Ascites and peripheral edema are observed with the development of biliary cirrhosis and portal hypertension (see Chapter 70A). These findings are less common today, because PSC patients are usually evaluated before the development of end-stage liver disease. A commonly encountered clinical scenario is a patient with CUC who presents with a cholestatic pattern of liver enzymes. Further medical evaluation results in the concurrent diagnosis of PSC. As with other chronic liver diseases, health-related quality of life is significantly impaired among PSC patients compared with healthy individuals (Younossi et al, 2001).
Diagnostic Criteria
The diagnosis of PSC is made based on clinical presentation, biochemical profile, and characteristic cholangiographic appearance of the bile ducts. Before the 1960s, PSC was a diagnosis of exclusion following abdominal exploration and biopsy of a suspected bile duct neoplasm (Schwartz & Dale, 1958). In the 1960s, diagnosing PSC became more frequent, when its association with IBD was recognized (Thorpe et al, 1967). With the advent of endoscopic retrograde cholangiopancreatography (ERCP) and percutaneous transhepatic cholangiography (PTC) in the 1970s, the characteristic “beaded” or “pruned tree” cholangiographic appearance obviated the need for exploratory surgery to diagnose PSC and resulted in its increased recognition (Wiesner & LaRusso, 1980; see Chapter 18).
The first formal diagnostic criteria included 1) absence of previous operative trauma to the biliary system; 2) sclerosis and stenosis involving all or most of the extrahepatic bile ducts; 3) exclusion of malignant disease involving the biliary tree, such as cholangiocarcinoma; and 4) absence of calculi in the gallbladder and common bile duct (Holubitsky & McKenzie, 1964). Following the introduction of ERCP, the criteria were expanded to include a subset of PSC cases recognized to have intrahepatic bile duct involvement alone (MacCarty et al, 1983). In addition, serial ERCP examinations of PSC patients demonstrated the subsequent development of cholangiocarcinoma (Chapman et al, 1980). As such, malignancy arising in the setting of sclerosing cholangitis was eliminated as an exclusionary criterion. Imaging techniques (i.e., ERCP) have also shown biliary tract calculi in many patients with established PSC, suggesting the removal of calculi as an exclusionary criterion (Kaw et al, 1995).
The diagnosis of PSC currently relies on 1) characteristic cholangiographic abnormalities of the biliary tree; 2) compatible clinical and biochemical findings, typically of ductal cholestasis with elevated serum alkaline phosphatase level for at least 6 months duration; and 3) exclusion of other causes of secondary sclerosing cholangitis (SSC). The clinical and cholangiographic features of SSC can mimic PSC, but SSC originates from known pathologic processes. Several well-described causes of SSC are listed in Table 41.1 (Abdalian & Heathcote, 2006). Currently, the most common presentation is an asymptomatic patient with persistently elevated levels of alkaline phosphatase first noted on routine serum biochemical screening. In the proper clinical context, a characteristic cholangiographic appearance of the bile ducts is sufficient for making the diagnosis.
Table 41.1 Causes of Secondary Sclerosing Cholangitis
AIDS, acquired immunodeficiency syndrome
Liver biopsy is not always necessary for diagnosing PSC, nor does it contribute new information that affects the clinical management of patients. In a study of 79 patients with PSC who had an already established diagnosis by cholangiography, liver biopsy did not affect management (Burak et al, 2003). Thus the role of liver biopsy in PSC is to 1) exclude other causes of liver disease, 2) diagnose small-duct PSC (discussed later), and 3) define the disease stage for determining prognosis and assessing efficacy of treatment prior to entering in therapeutic trials. Small-duct PSC comprises approximately 5% of histologically confirmed PSC cases (Angulo et al, 2002) and has significantly better long-term prognosis compared with classic PSC. However, some patients with small-duct disease can progress to classic PSC (Angulo et al, 2002). Patients with small-duct PSC usually have IBD and are seen intially with a cholestatic pattern of liver enzymes but normal cholangiography. Nevertheless, liver biopsy reveals classic histologic features of PSC. In the majority of patients, the history, serum biochemical profile, and cholangiography distinguish PSC from other causes of chronic cholestatic liver disease. The differential diagnosis of PSC is presented in Table 41.2.
Table 41.2 Differential Diagnosis of Primary Sclerosing Cholangitis
Biochemical and Serologic Abnormalities
Biochemical cholestasis of at least 6 months’ duration gives reason to suspect PSC. Alkaline phosphatase remains the most commonly elevated liver enzyme, often showing a threefold to fourfold increase. Nevertheless, a normal alkaline phosphatase level does not exclude the diagnosis of PSC, because normal levels have been reported in patients with cholangiographically proven disease (Balasubramanian et al, 1988). Therefore a normal alkaline phosphatase level should not dissuade further investigation if the clinical history (e.g., presence of IBD) and other evidence suggest liver disease.
Aminotransferase levels are often modestly elevated in patients with PSC, except in children, in whom these levels can be markedly increased. On average, adults with PSC have aminotransferase levels less than three times the upper limit of normal (Lee & Kaplan, 1995). PSC patients with highly elevated aminotransferases may show concomitant serologic evidence and histologic features of AIH (Czaja, 1998).
Serum bilirubin levels are normal in 60% of patients at diagnosis (Talwalkar & Lindor, 2005), but bilirubin levels will markedly rise as PSC progresses. An abrupt, sustained rise of bilirubin may herald a dominant biliary stricture, a bile duct stone, or the development of cholangiocarcinoma; therefore it should prompt additional investigation. Serum copper and ceruloplasmin levels and hepatic and urinary copper values are often abnormal. Hepatic copper levels can be elevated to the degree seen in Wilson disease and primary biliary cholangitis (PBC) (LaRusso et al, 1984); the copper increase is a reflection of prolonged cholestasis.
No autoantibody is pathognomonic for diagnosing PSC. The prevalence of antineutrophil cytoplasmic antibodies (ANCAs), anticardiolipin antibodies, and antinuclear antibodies (ANAs) in patients with PSC is 84%, 66%, and 53%, respectively (Angulo et al, 2000). Antimitochondrial antibodies (AMAs) and anti–smooth muscle antibodies are rare in patients with PSC. Approximately 25% of patients have hypergammaglobulinemia, and immunoglobulin M (IgM) levels are the most commonly elevated component (Wiesner & LaRusso, 1980). Autoantibody testing is helpful to identify those PSC patients with concurrent AIH, but antibody titers are not important in following PSC activity.
Imaging Studies
Visualization of the biliary tree is essential for establishing the diagnosis of PSC, and ERCP is the gold standard imaging technique (see Chapter 18). Typical cholangiographic findings of PSC include multifocal stricturing and beading throughout the biliary tree characteristic of alternating fibrosis and ectasia of the bile ducts (Fig. 41.1). Strictures of the bile ducts are the hallmark of the disease. They are often diffusely distributed and annular with intervening segments of dilated ducts. The classic cholangiographic findings involve both the intrahepatic and extrahepatic biliary tree. Strictures can vary in length and confluency, ranging from a bandlike appearance 1 to 2 mm in length to dense, annular strictures several centimeters in length. Approximately 25% of patients have biliary outpouchings that resemble diverticula. Similarly, 30% to 40% of patients may have mural irregularities that produce a shaggy appearance, varying from a fine brush border to frank nodularity (MacCarty et al, 1983).
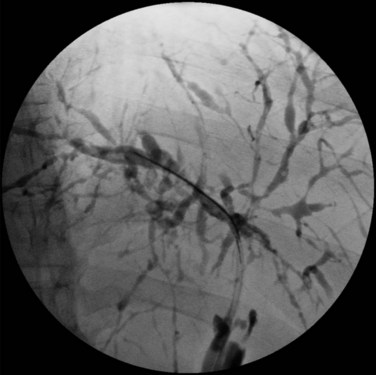
In a study of 86 patients with PSC, cholangiographic findings revealed involvement of the intrahepatic and extrahepatic ducts in 80 and 85 patients, respectively, when there was adequate visualization of the intrahepatic biliary tree. Involvement of only the intrahepatic and proximal extrahepatic ducts was seen in 20% of patients, and a very small number of patients had only small-duct PSC with a normal cholangiogram (MacCarty et al, 1983). Gallbladder and cystic duct involvement is less severe but is present in as many as 15% of patients (Brandt et al, 1988). ERCP should be considered in all patients suspected of having PSC. In addition to establishing the diagnosis, ERCP defines the extent and distribution of disease, identifies benign dominant strictures for endoscopic dilation or stenting, and allows brushings for cytology studies to screen for cholangiocarcinoma.
As many as 10% of cholangiocarcinomas have radiologic features that mimic PSC. Cholangiographic features that may suggest malignant degeneration of established PSC are markedly dilated biliary ducts or ductal segments, presence of a polypoid mass of 1 cm or greater in diameter, and progressive stricture formation. Nevertheless, these cholangiographic features are not specific for cholangiocarcinoma and can be seen in the absence of malignancy (MacCarty et al, 1985).
Despite the high diagnostic and therapeutic capabilities of ERCP, the procedure is invasive and is associated with clinical complications. The Mayo Clinic experience has shown that among PSC patients undergoing ERCP, the estimated rate of procedural complications requiring hospitalization is more than 10%. PSC patients undergoing ERCP are expected to have longer procedural time and a higher incidence of cholangitis compared with non-PSC patients (see Chapter 43), but the risk of pancreatitis, perforation, and bleeding are similar (Bangarulingam et al, 2009).
Magnetic resonance cholangiography (MRC) is a noninvasive substitute for ERCP (Fig. 41.2; see Chapter 17). Recent studies have estimated the sensitivity and specificity of MRC for diagnosing PSC to be between 80% to 82% and 87% to 98%, respectively (Talwalkar et al, 2004; Berstad et al, 2006). These studies concluded that MRC has diagnostic accuracy comparable to ERCP and results in cost savings when used as the initial diagnostic strategy. In another study, MRC was shown to be better than ERCP in identifying peripheral intrahepatic bile duct strictures (Vitellas et al, 2002).
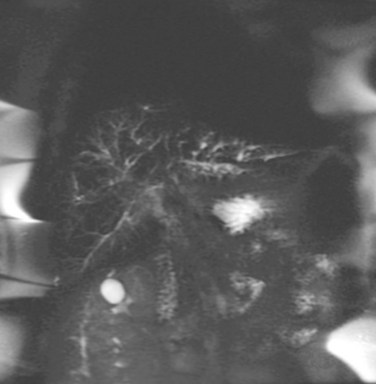
In a retrospective, nonblinded study of PSC patients with known biliary tract carcinoma, MRC findings of a well-defined mass exhibiting abnormal signal intensity were classified as definite cholangiocarcinoma in six or seven patients (Vitellas et al, 2002). MRC will likely replace diagnostic ERCP, but at present it is less sensitive and does not allow for biliary biopsy and cytology or therapeutic intervention.
PTC is useful when access to the biliary tree by ERCP is not technically feasible. Abdominal ultrasonography (US) and computed tomography (CT) are valuable in affirming ductal dilation and evaluating complications of PSC such as biliary stones, cirrhosis, and bile duct malignancy (see Chapters 13 and 16). US is often able to show large duct dilation and mural thickening of the common bile duct in patients with confirmed abnormalities on ERCP. It can further be used to detect gallstones or biliary tract calculi, which are important in the evaluation of bacterial cholangitis, worsening cholestasis, or jaundice.
Morphologic features of ductal abnormalities and cirrhosis can be seen on CT. Atrophy of the left lateral segment and hypertrophy of the caudate lobe may differentiate cirrhosis associated with PSC from that seen in other types of cirrhosis (Caldwell et al, 2001). CT can also complement cholangiography in the evaluation of malignancy with its ability to detect peripheral intrahepatic cholangiocarcinoma and metastatic spread within the liver parenchyma and the abdomen (Campbell et al, 1998, 2001). However, perihilar lymphadenopathy is very common in PSC with or without cholangiocarcinoma, and this finding cannot be taken as direct evidence of malignancy or metastasis.
Pathology
PSC can affect the entire biliary system. On rare occasions, it may be limited to the small intrahepatic ducts and be detectable only on histology. Small-duct PSC is thought to account for 5% of histologically confirmed PSC cases, although its true prevalence remains unknown (Angulo et al, 2002).
Liver biopsy findings alone are rarely sufficient to establish the diagnosis of PSC, which is characterized by damage to, atrophy of, and ultimately loss of bile ducts, thus sharing histologic features with other hepatobiliary diseases (Scheuer, 1998). Liver biopsy specimens from patients show portal tract inflammation and sclerosis (Fig. 41.3). Afflicted bile ducts are surrounded by a cuff of lightly inflamed sheets of fibrous tissue and edema, causing the fibrotic layers to separate and form the characteristic onion skin appearance. This classic histologic finding is nearly pathognomonic, but it is seen in fewer than 10% of PSC patients (Ludwig et al, 1981). Affected bile ducts will eventually atrophy and be replaced by rounded scars.
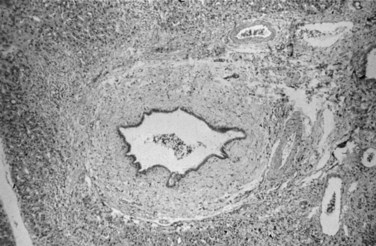
On occasion, similar fibroobliterative cholangitis can be seen in 1) PBC, 2) mechanical obstruction of larger bile ducts, 3) ductopenic rejection following liver transplantation, and 4) after intraarterial infusion of 5-fluorouracil. Involvement of the large intrahepatic and extrahepatic ducts distinguishes PSC from PBC. Inflammation in early-stage PSC is generally milder than that of PBC, but this distinction is difficult to make (Scheuer, 1998). Granulomas, thought to be classically associated with PBC, may be seen in about 4% of biopsies from PSC patients, making the distinction between PSC and PBC even more challenging (Ludwig et al, 1995).
Distinguishing between PSC-related histopathologic features and changes as a result of simple obstruction of distal bile ducts makes the liver biopsy interpretation difficult. Canalicular cholestasis is nonspecific and can occur as a result of any cause of biliary obstruction. As PSC progresses, however, the histopathologic changes of chronic cholestasis spill into the hepatic parenchyma. The most commonly used liver histology grading system proposed by Ludwig is based on these parenchymal changes. Disease was designated as stage 1, portal; stage 2, periportal; stage 3, septal; and stage 4, cirrhotic (Table 41.3; Ludwig et al, 1986). Histologic changes in the same liver can be markedly varied from segment to segment at any given point in time. Whereas histologic staging was formerly used as an independent predictor of the disease’s natural history, the revised PSC survival model does not take into account the histologic stage (Kim et al, 2000).
Table 41.3 Staging of Primary Sclerosing Cholangitis
| Portal stage (stage I) | Portal edema, inflammation, ductal proliferation; abnormalities do not extend beyond the limiting plate |
| Periportal stage (stage II) | Periportal fibrosis and inflammation with or without ductal proliferation; piecemeal necrosis may be present |
| Septal stage (stage III) | Septal fibrosis or bridging necrosis can be identified |
| Cirrhotic stage (stage IV) | Biliary cirrhosis evident |
Etiopathogenesis
The strong association of PSC with IBD has drawn much attention to the potential role of the inflamed colon. The hypothesis suggests that inflammation of the colon may increase permeability to various intraluminal products—toxins, bacteria, inflammatory mediators—that ultimately lead to liver disease. Bacteria or their toxic metabolic products have been considered but have not been conclusively shown to have a pathogenetic role in PSC development (Eade & Brooke, 1969; Palmer et al, 1980). However, investigators have shown that the absence of low-grade, chronic portal vein bacteremia in patients with CUC could potentially elicit the immune response that is responsible for PSC (Warren et al, 1966). Similarly, histologic evidence of bacteremia manifested as portal phlebitis is mild or absent in most PSC patients (Ludwig et al, 1981). Additional information showed that colonic bacteria might metabolize primary bile acids into toxic bile salts that are then absorbed through the ulcerated colon into the portal circulation, but no evidence of bile acid metabolism abnormalities in patients with PSC or CUC has been found (Siegel et al, 1977).
Animal models of PSC have reported that bacterial chemotactic peptides can lead to portal inflammation and histologic changes of PSC (Hobson et al, 1988: Lichtman et al, 1990). Suppression of the inflammatory response was reported in an animal model following inhibition of tumor necrosis factor (TNF). Nevertheless, in a clinical trial of PSC patients with pentoxifylline, a TNF inhibitor, no beneficial effect on symptoms or liver tests was seen (Bharucha et al, 2000). Clinical observations also cast doubt upon the role of the inflamed colonic mucosa in the development of PSC. First, PSC develops in approximately 25% of patients without evidence of IBD. Invasive screening of asymptomatic patients with PSC reveals that many have no endoscopic or histologic evidence of IBD. Second, lack of association between the severity of colonic disease and the likelihood of development and severity of PSC strengthens the skepticism that CUC may not directly cause PSC. Third, failure of proctocolectomy to modify the natural history of PSC argues against a direct causative role of CUC in PSC (Cangemi et al, 1989; Steckman et al, 1984). Thus, the putative contribution of increased colonic permeability in CUC resulting in inflammation of bile ducts remains unclear.
Genetic factors seem to play a role in the development of PSC. First, there have been reports of familial PSC among siblings (Quigley et al, 1983; Jorge et al, 1987). Second, various HLA associations with PSC have been reported, and HLA B8, DR3, DR2, and haplotype A1, B8, and DR3 have been found more frequently in patients with PSC compared with controls (Chapman et al, 1983; Donaldson et al, 1991; Schrumpf et al, 1982; Wiencke et al, 2007). PSC associated with DR4 is a more aggressive disease, although this correlation has not been universally found (Mehal et al, 1994; Olerup et al, 1995). Additional associations with DRB3*0101, DRB1*0301, DQA1*0501, DQB1*0201 and DRB1*1301, and DQA1*0103 and DQB1*0603 have been reported (Donaldson & Norris, 2002). Polymorphisms in the TNF-α receptor have been described as a possible genetic link to PSC. G to A substitution at position −308 in the TNF-α gene has been associated with susceptibility to PSC (Mitchell et al, 2001). A functional variant of stromelysin, matrix metalloproteinase 3, may also influence PSC susceptibility and disease progression (Satsangi et al, 2001). Moreover, variations in the MICA gene (major histocompatibility complex class I–related MIC gene family) have a role in PSC predisposition. Independent of other HLA haplotypes, the MICA 002 allele appears to significantly reduce the risk of PSC, and the MICA 008 allele increases the risk of developing PSC (Norris et al, 2001). Finally, the CCR5-δ32 mutation, characterized by a 32-base pair deletion in the CCR5 gene of T cells, has been associated with susceptibility to PSC development and severity (Eri et al, 2004).
Immune-mediated damage to cholangiocytes seems to be the most likely mechanism leading to PSC. Theoretically, certain HLA molecules and haplotypes may contribute by eliciting an immune response against antigenic epitopes present on biliary epithelia. Enhanced expression of MHC class II antigens (i.e., HLA DR) on cholangiocytes in early-stage PSC has drawn suspicion regarding its role in altered immunity in disease pathogenesis. Aberrant expression of HLA DR is also apparent in PBC and extrahepatic biliary obstruction, suggesting that expression of this antigen is an epiphenomenon rather than an implicit cause of PSC (Broome et al, 1990; Chapman et al, 1988; Van Milligen De Wit et al, 1995).
Decreased hepatic clearance of circulating immune complexes, generalized complement activation, and sharing of a specific epitope between human colonic and biliary epithelial cells have also been reported (Bodenheimer et al, 1983; Minuk et al, 1985; Das et al, 1990; Mandal et al, 1994), yet none of these observations have proven pathogenic associations with PSC. Enhanced interactions of intracellular adhesion molecule 1 (ICAM1) present on cholangiocytes with its cognate ligand on T lymphocytes, leukocyte function–associated antigen 1 (ITGAL), may play a role in PSC development. Genetic polymorphisms of ICAM1 have been implicated in susceptibility to PSC. Homozygote status of the E469E allele for ICAM1 has been associated with protection against PSC (Yang et al, 2004). Both enhanced expression on proliferating cholangiocytes and increased serum levels of ICAM1 have been reported in PSC patients (Van Milligen De Wit et al, 1995; Adams et al, 1991; Bloom et al, 1995).
Associated Diseases
PSC is strongly associated with IBD, most commonly CUC. PSC patients may also have biochemical, serologic, and histologic features of AIH, and a variety of other diseases have been reported to associate with PSC. Given the lack of large studies, it is unclear whether these weak associations are true or simply coincidental. An abridged list of associated diseases is presented in Table 41.4.
Table 41.4 Diseases Associated with Primary Sclerosing Cholangitis
Inflammatory Bowel Diseases
The close association of PSC with IBD, particularly CUC, is widely recognized. IBD is seen in approximately 70% to 80% of patients with PSC, and CUC accounts for 85% to 90% of those patients; Crohn disease is responsible for the rest (Fausa et al, 1991; Loftus et al, 1997). Patients with PSC and Crohn disease may have milder liver disease than patients with PSC and CUC, and PSC has not been seen in Crohn disease involving only the small intestine (Rasmussen et al, 1997). Typically, the diagnosis of IBD is established 8 to 10 years before the liver disease is evident, although there is no clear temporal association, and cases of IBD occurring years after diagnosis of PSC have also been reported (Chapman et al, 1980; Loftus et al, 1996a, 1998). No direct correlation has been found between the severity of bowel disease and the severity of liver disease. Furthermore, therapy of the IBD does not alter the course of the liver disease. For instance, proctocolectomy, the most aggressive treatment for CUC, has no effect on PSC natural history (Cangemi et al, 1989). Colitis is usually milder in patients with both CUC and PSC in comparison to patients with CUC alone.
PSC may play a role in the development of colorectal dysplasia in the setting of CUC, as patients with CUC and PSC show a higher risk of colonic dysplasia compared with patients who have only CUC (Broome et al, 1995a, 1995b). In a study from Sweden, the absolute cumulative risk of developing colorectal dysplasia/carcinoma in patients with PSC and CUC was 9%, 31%, and 50% after 10, 20, and 25 years of disease duration, respectively. In patients with CUC alone, the parallel risk was 2%, 5%, and 10%, respectively (Broome et al, 1995b). Subsequent studies also confirmed the observation of Broom and others, where predicted risk of colorectal carcinoma with PSC and concomitant IBD after 10 years of disease duration was up to 25% (Kornfeld et al, 1997; Claessen et al, 2009). Other investigators have reported that patients with both PSC and CUC were five times more likely to develop colonic dysplasia, based on mucosal biopsy, compared with patients with CUC alone (Brentnall et al, 1996
Related posts:
 Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
 Nonhepatic surgery in the cirrhotic patient
Nonhepatic surgery in the cirrhotic patient
 Liver transplantation for cholangiocarcinoma and other neoplastic diseases
Liver transplantation for cholangiocarcinoma and other neoplastic diseases
 Distal splenorenal shunt
Distal splenorenal shunt
 Interventional endoscopy: Technical aspects
Interventional endoscopy: Technical aspects
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


