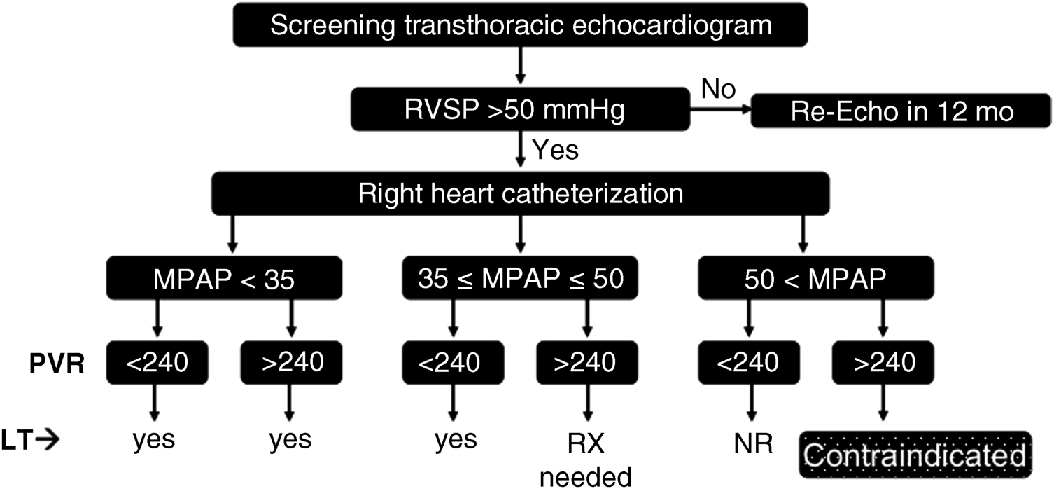
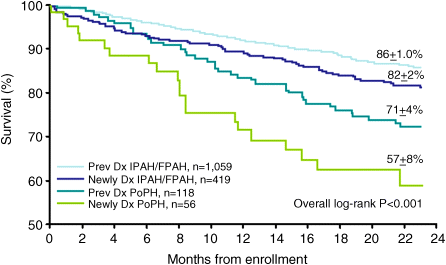
Chapter 19 Rodrigo Cartin-Ceba and Michael J. Krowka Division of Pulmonary and Critical Care Medicine, Mayo Clinic, Rochester, MN, USA Portopulmonary hypertension (PPH) is the well-recognized relationship of pulmonary arterial hypertension (PAH) that evolves as a consequence of portal hypertension [1]. PPH is included within Group I of the 5th World symposium on pulmonary hypertension [2]. The first clinical and pathologic report of what we now know as PPH was provided by Mantz and Craige in 1951 [3]. These authors described necropsy results of a 53-year-old female with spontaneous portocaval shunt (due to a probable congenital portal vein narrowing) that originated at the confluence of the portal, splenic, and mesenteric veins, coursed through to mediastinum and was lined by varying amounts of thrombus thought to have embolized via the innominate vein into the right heart and pulmonary arteries. In addition to embolized small pulmonary arteries, an extreme endothelial proliferation and recanalization process was documented [3]. Since the 1980s, enhanced recognition and renewed importance of PPH has evolved with the evolution of liver transplantation (LT) and potential outcomes associated with PPH. Specific screening recommendations and diagnostic criteria are now clearly defined for this syndrome. Despite the lack of randomized controlled trials for its medical treatment, extrapolation of the therapeutic advances in treating pulmonary artery hypertension with specific effects in PPH has stimulated ongoing interest and importance in this syndrome. This chapter summarizes the most recent advances in the comprehensive management of patients with PPH. PPH is only one of the various pulmonary hemodynamic patterns that complicate advanced liver disease [4,5] and therefore the importance of accurate interpretation of hemodynamics obtained by right heart catheterization (RHC) is vital in order to provide appropriate management. The vascular pathology that characterizes PPH includes obstruction to arterial flow due to vasoconstriction, endothelial and smooth muscle proliferation, in situ thrombosis, and plexogenic arteriopathy [6,7]; these changes increase the resistance to pulmonary arterial blood flow which is the main mechanism of the disease. PPH is defined as a mean pulmonary artery pressure (MPAP) ≥25 mmHg associated with pulmonary vascular resistance (PVR) ≥240 dyn/s/cm−5 and pulmonary artery wedge pressure (PAWP) <15 mmHg based upon RHC. A summary of the diagnostic criteria and severity classification of PPH is presented in Table 19.1. It is also very important to recognize the three main abnormal hemodynamic patterns that can be present during RHC in patients with portal hypertension (Figure 19.1): Table 19.1 Diagnostic and severity criteria for portopulmonary hypertension. Figure 19.1 Pulmonary hemodynamic patterns documented by right heart catheterization in advanced liver disease. CO, cardiac output; MPAP, mean pulmonary artery pressure; PAWP, pulmonary artery wedge pressure; PPH, portopulmonary hypertension; PVR, pulmonary vascular resistance. Distinguishing these patterns is of paramount important for adequate management and treatment. Transthoracic echocardiography (TTE) has been the most practical screening method to detect PPH [8–10]. By assessing the tricuspid regurgitant peak velocity (TR), estimating the right atrial pressure by inferior vena cava changes with inspiration and using the modified Bernoulli equation, an estimate of right ventricle systolic pressure (RVSP) can be determined in ∼80% of patients with portal hypertension [8]. This quantitative approach allows one to decide which patients should proceed to RHC for the definitive characterization of pulmonary hemodynamics. The presence of RVSP >50 mmHg has been the cutoff criterion to proceed to RHC in the current Mayo Clinic algorithm followed since 1996 [5]; rarely, immeasurable TR with abnormal qualitative right ventricular size or function results in RHC. TTE was noted to have a 97% sensitivity and 77% specificity to detect moderate to severe PAH prior to LT [8]. Identifying which patients indeed have PPH, deciding who needs PAH-specific therapy based on severity, and determining the risks and timing for potential LT are clinical issues based upon RHC results and are summarized in Figure 19.2. Those PPH patients with MPAP >35 mmHg are particularly vulnerable to poor outcomes with attempted LT, especially if there is no attempt to treat the PPH with current PAH-specific therapy [11]. With current treatments, PPH outcomes are variable, yet in highly selected PPH patients, with aggressive treatment and successful LT, pulmonary hemodynamics may completely normalize. Right ventricular size and function normalizes and liberation from PAH-specific therapy may be allowed. Figure 19.2 Current PPH screening evaluation and treatment algorithm used at the Mayo Clinic. Contraindicated, high risk of intraoperative event at graft reperfusion; LT, liver transplantation; MPAP, mean pulmonary artery pressure (normal <25 mmHg); NR, never reported; PVR, pulmonary vascular resistance [normal <240 dyn/s/cm−5 (or 3 Wood units)]; RVSP, right ventricular systolic pressure estimated by transthoracic echocardiography; RX, vasomodulating treatment advised prior to LT; Yes, risk to proceed with liver transplant is minimal. Importantly, PPH should be distinguished from the other major pulmonary vascular consequence of liver disease, hepatopulmonary syndrome (HPS) [4,11]. In HPS, arterial hypoxemia is caused by intrapulmonary vascular dilatations (exactly opposite to the vascular obstructions documented in PPH) that form as a remodeling process due to factors yet to be identified. In addition, the pulmonary hemodynamics associated with HPS reflect a normal pulmonary vascular resistance and usually a high flow state characterized by an increased cardiac output (CO). The distinction between these two syndromes is summarized in Table 19.2 and is quite important especially if LT is to be considered because of the differences in risk and outcomes between these two syndromes [11]. Table 19.2 Distinctions between portopulmonary hypertension (PPH) and hepatopulmonary syndrome (HPS). It is unclear as to who coined the term “portopulmonary hypertension”, but Yoshida et al. [12] appear to be the first authors to use the term in 1993 as they described the first successful case of PPH to undergo successful LT (39-year-old male with long-standing chronic active hepatitis). Subsequently, several small series and case reports with autopsy results have described pulmonary arterial obstruction and pulmonary plexogenic arteriopathy with and without thromboemboli [7,13–16]. Two distinct pulmonary vascular obstructive patterns causing PAH in association with portal hypertension are well described: Large series have confirmed the coexistence of these portal and pulmonary vascular abnormalities and have shown that they are not coincidental. An unselected series of 17 901 patients documented PAH associated with cirrhosis more than five times more frequently than PAH without cirrhosis [17]. Within the 1981–1987 NIH national registry of “primary” pulmonary hypertension from 32 centers reported by Rich et al. [18], additional analyses by Groves et al. [19] concluded that 8.3% likely had PPH (17/204; 187 had primary pulmonary hypertension). Hadengue et al. [20] reported the largest prospective study of patients with portal hypertension (n = 507) in which portopulmonary hemodynamic measurements concluded that 2% had PPH. With the era of PAH-specific therapy and LT, prospective studies have focused on the frequency of PPH in clinical settings, national registries, and individual transplant center experiences. Humbert et al. [21] reported a 10.4% frequency of PPH (70/674) from 17 French university hospitals pulmonary hypertension registry clinic experiences over a 12-month period (2002–2003). The US-based REVEAL Registry documented a 5.3% PPH frequency (174/3; 525) in which there were 68% prevalent and 32% incident cases satisfying the criteria that MPAP >25 mmHg and PVR >240 dyn/s/cm−5 with PAWP <15 mmHg [22]. Of interest were the additional 29 patients that satisfied MPAP and PVR criteria, but in whom the PAWP was >15 mmHg; each of those patients had a transpulmonary gradient >12 mmHg (abnormal). Following slightly different PVR diagnostic criteria as part of outpatient RHC diagnostic assessments, the largest PPH-liver transplant center experiences reported to date are as follows: 8.5% (Baylor 102/1205; PVR >120 dyn/s/cm−5), 6.1% (Clichy, France 10/165; PVR >120 dyn/s/cm−5), and 5.3% (Mayo Clinic 66/1235; PVR >240 dyn/s/cm−5) [5,23,24]. The natural history of PPH has been difficult to characterize and has also been confounded by small series from eras in which none of the current PAH-specific therapies were available versus the current era with the evolving PAH-specific therapies and LT experiences. Robalino and Moodie [25] reported a dismal 5-year survival rate of 4% (n = 78) in an era prior to the availability of continuous IV prostacyclin infusion. Swanson et al. [26] reported a 14% 5-year survival rate in PPH patients (n = 19) denied LT and not treated with any of the current PAH-specific therapies. From the French National Center for PAH (n = 154 over a 20-year span until 2004), Le Pavec et al. [27] described 1, 3, and 5-year survival rates of 88%, 75%, and 68%, respectively, PPH patients (but mainly Child A and alcoholic cirrhotics). Only one-third had been treated with PAH-specific therapies with severity of cirrhosis and reduced CO poor prognostic factors. Causes of death in all series mentioned herein were equally distributed between right heart failure due to PPH and direct complications of liver disease (bleeding, sepsis, hepatocellular carcinoma). From the REVEAL registry (Registry to Evaluate Early And Long-term pulmonary arterial hypertension disease management) two important PPH observations were reported [22]. First, PPH treatment patterns reported in the REVEAL registry demonstrated that the use of any PAH-specific therapy for PPH was delayed compared with patients diagnosed with idiopathic pulmonary artery hypertension (IPAH). Specifically, at the time of entry into the registry only 25% were on PAH-specific therapy; by the end of 12 months’ follow-up 74% of those alive were on treatment. Second, although baseline hemodynamics in PPH (MPAP and PVR) were significantly better than those with IPAH, the 1 and 3-year survival rates were worse (Figure 19.3); the 5-year survival rate for all PPH patients was 40% versus 64% for IPAH. Liver disease etiologies and causes of death were not determined in the registry and survival was not analyzed by the type of PAH-specific therapy. Figure 19.3 Two-year survival patterns for portopulmonary hypertension (PPH) and idiopathic pulmonary artery hypertension (IPAH) categorized by previous versus newly diagnosed at the time of entry into the registry. FPAH, familial pulmonary arterial hypertension. (Krowka et al., 2012 [22]. Reproduced with permission of Chest.) Two caveats are important in characterizing the natural history of PPH. First, aside from one study evaluating the effect of riociguat (a soluble guanylate cyclase stimulator) in PAH [28], controlled randomized studies evaluating PAH-specific therapies have excluded PPH patients. This universal exclusion in the United States further complicated the understanding of PPH outcomes compared to other PAH disorders. Second, beginning in 2002, a higher priority for LT was an option for highly selected patients with PPH in the United States [29]. Formalization of higher priority pulmonary hemodynamic criteria were put forth in 2006 and standardized in 2010. Only patients with moderate to severe PPH (MPAP >35 mmHg) who attained significant hemodynamic improvement with PAH-specific therapy (MPAP <35 mmHg and PVR <400 dyn/s/cm−5) were granted higher priority for LT. From 2002 through 2010, 155 PPH patients were granted such priority and transplanted by regional review boards [30]. It is not clear why only 6–8% of patients with portal hypertension develop PPH, which is indistinguishable from the histopathology of other PAH phenotypes [6,7]. Based upon autopsy and lung explant studies, PPH is characterized by a spectrum of obstructive and remodeling changes in the pulmonary arterial bed. Initially, medial hypertrophy with smooth muscle proliferation and a transition to myofibroblasts has been documented. Intuitively, this vasoconstrictive process could be responsive to pulmonary arterial vasodilator therapy. As this proliferative pathologic process advances, platelet aggregates, in situ thrombosis, intimal fibrosis, and finally, taking a sporadic, web-like shape involving the entire pulmonary arterial wall with recanalization for the passage of pulmonary arterial blood [6,7]. This web-like pathology is termed plexogenic arteriopathy and it is unknown whether such structures are irreversible [7]. The factors leading to such pulmonary vascular obstruction in the setting of liver disease are complex, but originate from the development of portal hypertension. The pulmonary vascular pathology occurs within the context of a hyperdynamic state caused by extrahepatic (splanchnic) vasodilatation [31]. It is unknown if this persistent high flow state initiates (by shear stress) or exacerbates (in combination with circulating mediators) the pulmonary vascular proliferative process. Genetic predisposition may also have a role because not all patients with portal hypertension due to cirrhosis develop PPH [32]. It has been demonstrated that the pulmonary endothelial cells lack prostacyclin synthase in PPH patients (hence a lack of prostacyclin vasodilatation) [33], the pulmonary vascular bed is exposed to increased levels of circulating endothlin-1 in the setting of cirrhosis (a potent vasoconstrictor and facilitator of smooth muscle proliferation) [34,35], and may be deficient in local nitric oxide effect (for vasodilatation) [36]. The role of other circulating and receptor factors that may affect the pulmonary endothelium is speculative. These factors include vasoconstrictive/proliferative mediators such as serotonin, thromboxane, vasoactive intestinal peptide, and vascular endothelial growth factor (VEGF); as well as the possible imbalance of endothelin receptors (ETA – mediating vasoconstriction; ETB (mediating vasodilatation) in the pulmonary arterial bed [36]. The mechanistic link between estrogen signaling, serum estradiol levels, circulating endothelial progenitor cells, and the development of PPH is a current research hypothesis of interest [37,38]. It is also hypothesized that, initially, the intrahepatic injury leads to an intrahepatic increased resistance to flow. Such injury evokes mediators that cause splanchnic vasodilatation via enhanced local nitric oxide effect and presumed angiogenesis effect [31,39]. The splanchnic bed becomes hyporesponsive to vasoconstrictors; excess volume occurs due to renal retention and a hyperdynamic state is generated. This high flow exacerbates the existing portal hypertension due to increased intrahepatic resistance to flow, leading to the creation of alternative flow pathways: portocaval shunts and esophagogastric varices. Presumably, such aberrant pathways allow other mediators to reach the pulmonary arterial bed and, in the setting of high flow, “trigger” the pulmonary arterial proliferative process.
Portopulmonary Hypertension1
Introduction
Definition
Variable
Criterion
Portal hypertension
Clinical diagnosis (ascites, varices, splenomegaly)
Mean pulmonary artery pressure (MPAP)
≥25 mmHg; and
Pulmonary vascular resistance (PVR)a
>240 dyn/s/cm−5
Pulmonary artery wedge pressure (PAWP)
<15 mmHg
Transpulmonary gradientb (TPG)
>12 mmHg
Degree of severity
Mild
25 < MPAP < 35 mmHg
Moderate
35 ≤ MPAP ≤ 45 mmHg
Severe
45 mmHg ≤ MPAP
TPG (MPAP–PAWP) distinguishes between simple volume excess causing increased MPAP and the pulmonary artery vasculopathy that characterizes PPH.
aPVR = (MPAP–PCWP) × 80/cardiac output.
bIn the case where PAWP is >15 mmHg (abnormal), an abnormal.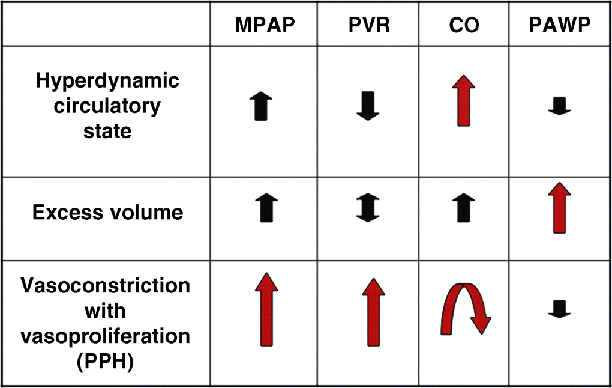
Screening
Variable
PPH
HPS
Presentation onset
Pediatric
Rare
Yes
Adult
Most common
Yes
Symptoms
Exertional dyspnea
Chest pain/syncope
Exertional dyspnea
Clinical exam
↑Second heart sound
Systolic murmur
Clubbing/cyanosis
Systolic murmur
Genetic predisposition
Yes
Unknown
Liver diseases
Autoimmune (common)
No relation
Portocaval shunts
Spontaneous (common)
No relation
Child–Turcotte–Pugh classes
No relation
No relation
MELD scores
No relation
No relation
Lung biopsies
Not indicated
Not indicated
Chest radiograph
Cardiomegaly
Enlarged central arteries
Normal
Electrocardiogram
RBBB
T wave inversion V1–V4
Normal
Transthoracic echo
↑ RV size; ↓ RV function
+ shunt
Normal RV
++++ shunt
Arterial hypoxemia
Mild
May be severe
Abnormal overnight oximetry
Common
Common
Pulmonary hemodynamics
↑MPAP; ↑PVR
↑MPAP; normal PVR
Pulmonary angiography
Enlarged central arteries
Distal pruning
Spongy appearance
Discrete AV communications (uncommon)
Liver transplant
MELD exception if MPAP, PVR improved
MELD exception if PaO2 < 60 mmHg
Causes of death
Right heart failure
GI complications
GI complications
GI, gastrointestinal; MELD, Model end-stage liver disease; MPAP, mean pulmonary artery pressure; PVR, pulmonary vascular resistance; RBBB, right bundle branch block; RV, right ventricle.
Epidemiology and Natural History
Etiology and Pathogenetic Mechanisms
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


