CHAPTER 90 Portal Hypertension and Gastrointestinal Bleeding
Variceal hemorrhage, hepatic encephalopathy, and ascites—the major complications of cirrhosis of the liver—result from portal hypertension, defined as an increase in hepatic sinusoidal pressure to 6 mm Hg or greater. Portosystemic collaterals decompress the hypertensive hepatic sinusoids and give rise to varices at the gastroesophageal junction and elsewhere. These portosystemic collaterals also may allow ammonia derived from the intestine to reach the brain, thereby resulting in hepatic encephalopathy through a pathologic process of several intermediary steps involving the peripheral benzodiazepine-type receptors, neurosteroids, and γ-aminobutyric acid (GABA) receptors (see Chapter 92). Additionally, portal hypertension is associated with renal retention of sodium and water and the formation of ascites (see Chapter 91). Indeed, portal hypertension and its complications remain important clinical problems despite advances in treatment and improved understanding of both the molecular basis and pathophysiology of portal hypertension.
NORMAL PORTAL CIRCULATION
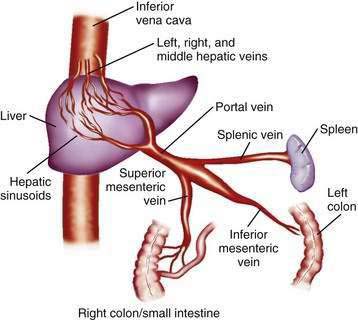
The portal venous system carries capillary blood from the esophagus, stomach, small and large intestine, pancreas, gallbladder, and spleen to the liver. The portal vein is formed by the confluence of the splenic vein and the superior mesenteric vein behind the neck of the pancreas.1 The inferior mesenteric vein usually drains into the splenic vein. The left gastric vein, also called the left coronary vein, usually drains into the portal vein at the confluence of the splenic vein and superior mesenteric vein (Fig. 90-1). The portal vein is approximately 7.5 cm in length and runs dorsal to the hepatic artery and bile duct into the hilum of the liver. The uppermost 5 cm of the portal vein does not receive any tributaries.2 In the hilum of the liver, the portal vein divides into the left and right portal vein branches, which supply the left and right sides of the liver, respectively. The umbilical vein drains into the left portal vein. The cystic vein from the gallbladder drains into the right portal vein, whereas the portal venules drain into hepatic sinusoids that, in turn, are drained by the hepatic veins into the inferior vena cava. The left and middle hepatic veins usually join and drain into the inferior vena cava separately but adjacent to the confluence of the right hepatic vein with the inferior vena cava. The caudate lobe drains separately into the inferior vena cava (see Chapter 71).
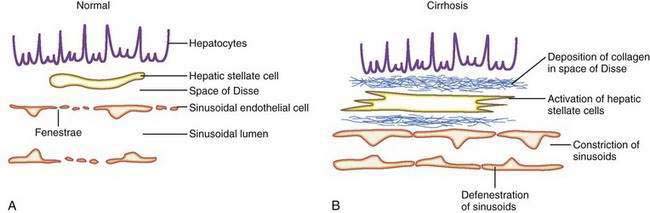
The sinusoids are highly permeable and thus facilitate the transport of macromolecules to the parenchymal hepatocytes that reside on the extraluminal side of the endothelial cells. The hepatic sinusoids are highly permeable because they lack a proper basement membrane and because the endothelial cells that line the sinusoids contain fenestrae. Other unique aspects of the hepatic sinusoids are the space of Disse, a virtual space located extraluminal to the endothelial cell and adjacent to the hepatocyte, and its cellular constituents, the hepatic stellate cell (HSC) and the Kupffer cell (Fig. 90-2; see also Chapters 71 and 72). These two cell types probably play an important role, in concert with the endothelial cell, in regulating sinusoidal hemodynamics and homeostasis and may contribute to the sinusoidal derangements that occur in portal hypertension. Under basal conditions, HSCs maintain a quiescent phenotype and accumulate vitamin A. On activation, however, as occurs in cirrhosis and portal hypertension, these cells are postulated to develop contractile abilities that permit them to function as sinusoidal pericytes. Kupffer cells contribute to vascular homeostasis by generating cytokines with potent cellular and vasoregulatory actions, including tumor necrosis factor. Endothelial cells and smooth muscle cells in nonsinusoidal hepatic vessels such as the portal venule and the terminal hepatic venule are important in hepatic vasoregulation, particularly in the normal liver, where HSCs are quiescent, unactivated, and presumably less contractile.
HEMODYNAMIC PRINCIPLES OF PORTAL HYPERTENSION
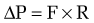
in which the pressure gradient in the portal circulation (ΔP) is a function of portal flow (F) and resistance to flow (R). Increases in portal resistance or portal flow can contribute to increased pressure. Portal hypertension almost always results from increases in both portal resistance and portal flow (Fig. 90-3). One exception is that of an arteriovenous fistula, which in the initial stages causes portal hypertension largely through an increase in portal flow in the absence of an increase in resistance. The mechanism of the increase in portal resistance depends on the site and cause of portal hypertension; in the Western world, the most common cause is liver cirrhosis (see later). Because of the increase in hepatic resistance and the decrease in hepatic compliance, small changes in flow that do not increase pressure in the normal liver can have a prominent stimulatory effect on portal pressure in the cirrhotic liver. The increase in portal venous inflow is part of a generalized systemic derangement termed the hyperdynamic circulatory state. Collateral vessels that dilate and new vascular sprouts that form connect the high-pressure portal venous system with lower-pressure systemic veins. Unfortunately, this process of angiogenesis and collateralization is insufficient for normalizing portal pressure and actually causes complications of portal hypertension, such as esophageal varices.3 Approaches to block this angiogenic process are a compelling target for drug development.
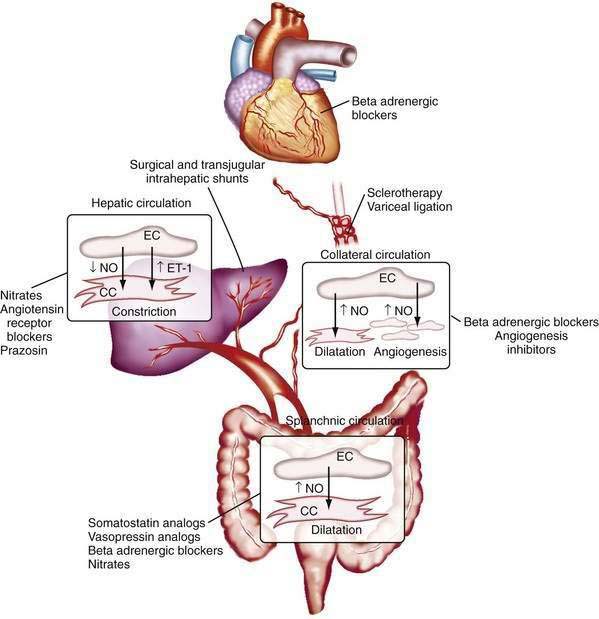
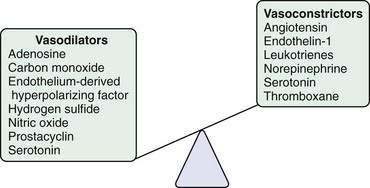
The changes in portal flow and resistance also can be viewed as originating from mechanical and vascular factors. Mechanical factors include the fibrosis and nodularity of the cirrhotic liver with distortion of the vascular architecture and the remodeling that is recognized to occur in the systemic and splanchnic vasculature in response to the chronic increases in flow and shear stress that characterize the hyperdynamic circulatory state. Vascular factors include intrahepatic vasoconstriction, which contributes to increased intrahepatic resistance, and the splanchnic and systemic vasodilatation that accompanies the hyperdynamic circulatory state. The vascular factors that contribute to portal hypertension are particularly important because they are reversible and dynamic and therefore compelling targets for experimental therapies (Fig. 90-4). Conversely, effective therapies for the fixed, mechanical component of portal hypertension caused by scar, regenerative nodules, and vascular remodeling are currently lacking. Indeed, most available therapies for portal hypertension focus on correction of hemodynamic alterations in the portal circulation. Approaches include use of nonselective β-adrenergic blocking agents, octreotide, and vasopressin to reduce the hyperdynamic circulation, portal venous inflow, and splanchnic vasodilatation.4,5 Alternative agents reduce the increased intrahepatic resistance and include angiotensin receptor blockers and mononitrates.
INCREASED INTRAHEPATIC RESISTANCE
In cirrhosis, increased portal resistance occurs in great part as a result of mechanical factors that reduce vessel diameter. In addition to regenerative nodules and fibrotic bands, these mechanical factors include capillarization of the sinusoids and swelling of cells, including hepatocytes and Kupffer cells. As discussed earlier, however, reduced hepatic vessel diameter resulting in increased portal resistance, even when caused by cirrhosis, is not a purely mechanical phenomenon.6 Hemodynamic changes in the hepatic circulation also contribute to increased intrahepatic resistance.7,8 These changes are characterized by hepatic vasoconstriction and impaired responses to vasodilatory stimuli. The increase in intrahepatic resistance is determined largely by changes in vessel radius, with small reductions in vessel radius causing prominent increases in resistance. Blood viscosity and vessel length also can influence resistance, albeit to a much smaller extent. The factors that regulate resistance can be viewed in the context of the law of Poiseuille:
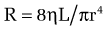
in which R is resistance, ηL is the product of blood viscosity and vessel length, and r is vessel radius.
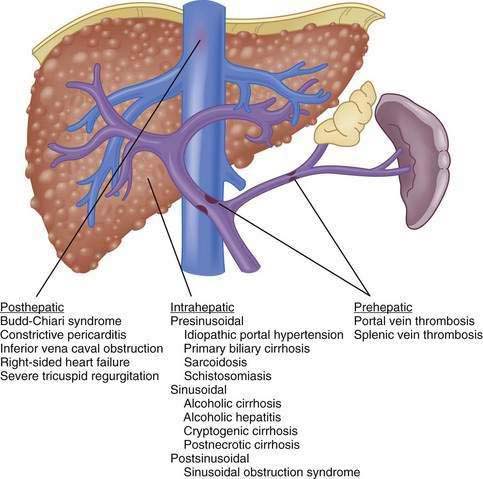
Although vasoactive changes were estimated initially to account for 10% to 30% of the increase in portal resistance in cirrhosis, subsequent studies have suggested that these figures actually may underestimate the contribution of hepatic vasoconstriction to the increased resistance observed in the cirrhotic liver. In noncirrhotic causes of portal hypertension, the increase in resistance may occur at sites upstream (prehepatic) or downstream (posthepatic) of the liver, as in portal vein thrombosis and hepatic vein thrombosis, respectively (Fig. 90-5). Furthermore, the site of increased intrahepatic resistance can be further delineated as the sinusoids (sinusoidal), upstream from the sinusoids within the portal venules (presinusoidal), or downstream from the sinusoids in the hepatic venules (postsinusoidal), as in alcoholic cirrhosis, schistosomiasis, and sinusoidal obstruction syndrome, respectively. Pressure is increased only in the portal circulation behind the site of increased resistance, and in isolated portal vein thrombosis, hepatic function frequently remains largely preserved despite prominent portal hypertension.
Most evidence suggests that a decrease in the production of the vasodilator NO and an increase in the production of the vasoconstrictor ET-1 jointly contribute to the increase in hepatic vascular resistance. In experimental models of cirrhosis, the bioavailability of hepatic NO is diminished because of a reduction in the production of NO by endothelial cells.7,9,10 A similar paradigm is observed in the human cirrhotic liver.11 Most studies indicate that the reduction in NO production occurs not through a reduction in hepatic eNOS protein levels9,10 but through defects in the steps necessary to activate existing eNOS protein. For example, increases in the production of the eNOS-inhibiting protein caveolin-1 have been observed in experimental models of cirrhosis10 and in human cirrhosis. Another pathway that contributes to deficient generation of NO by eNOS is a reduction in the level of AKT (protein kinase B) phosphorylation of eNOS and upregulation of the eNOS inhibiting protein, GRK (G protein-coupled receptor kinase), in the cirrhotic liver.12 Irrespective of the mechanism of deficiency, the lack of availability of NO is thought to allow HSCs, which are activated and highly contractile in liver cirrhosis, to constrict the sinusoids that they envelop, thereby increasing portal pressure. The role of the HSCs in this process remains controversial, however, because evidence is mixed regarding whether the site of the increase in intrahepatic resistance in cirrhosis is the sinusoids, where stellate cells reside, or the pre- or postsinusoidal venules (or both), which are devoid of stellate cells and in which endothelial cells signal smooth muscle cells. Furthermore, increasing evidence points toward diverse origins of these myofibroblastic cells within the cirrhotic sinusoids, with portal myofibroblasts as well as HSC postulated to play important roles.13 In this regard, therapies that target myofibroblast migration may be a compelling therapeutic target by limiting the density of these contractile cells within the hepatic sinusoids.14 Finally, the contribution of HSCs to hepatic angiogenesis may also be an important target for treating fibrosis and portal hypertension.15
In clinical practice, NO can be delivered by NO donor agents such as mononitrates. NO donor agents exert their beneficial effects in part by relaxing the actively contractile stellate cells.16,17 The systemic actions of these agents, however, tend to cause side effects and exacerbate the hyperdynamic circulatory state. In studies utilizing a liver-specific NO donor compound, the increased intrahepatic vascular resistance could be corrected by the generation of additional NO and consequent relaxation of HSCs.18 In cirrhosis, however, deficient endothelial cell NO generation may be accompanied by impaired stellate cell relaxation in response to NO,19 perhaps because of diminished response of the NO second messenger cyclic guanosine monophosphate (cGMP) in activated cells.16 In this situation, a prominent beneficial effect of NO donors is less predictable.
Excessive ET-1 also contributes to increased intrahepatic vasoconstriction in portal hypertension through vasoconstrictive effects in the liver, presumably by enhancing HSC contractility.20,21 In experimental models, ET-1 protein and receptor expression are increased, most notably in HSCs and endothelial cells.20,22,23 In humans with portal hypertension, plasma and liver ET-1 levels also are increased.24 The reason for activation of the ET-1 system in portal hypertension is not known, but this effect may be secondary to transforming growth factor-β (TGF-β), a key fibrogenic growth factor.23 Clinical trials of ET antagonists in patients with portal hypertension are in progress; however, the variable effects of ET modulation in experimental models of portal hypertension, as well as the possible hepatotoxicity of these compounds, have limited enthusiasm for studies in humans.25 Other therapies for portal hypertension may provide benefit through the ET pathway. For example, somatostatin, which reduces portal pressure by constricting the splanchnic circulation, also may act by inhibiting ET-1–dependent HSC contraction.26
Other vasoactive mediators, including cysteinyl leukotrienes, thromboxane, angiotensin, and hydrogen sulfide, also have been implicated in the development of increased intrahepatic resistance in cirrhosis.27,28 Some of these mediators, particularly angiotensin, which causes contraction of HSCs, have been studied in humans. Attempts to reduce portal pressure using pharmacologic agents that inhibit angiotensin activation of HSC contraction have met with mixed results thus far.29
HYPERDYNAMIC CIRCULATION
In addition to the increases in portal resistance discussed earlier, a major factor in the development and perpetuation of portal hypertension is an increase in portal venous flow, or the hyperdynamic circulation. The term portal venous inflow indicates the total blood that drains into the portal circulation, not the blood flow in the portal vein itself, which may actually be diminished in portal hypertension because of portosystemic collateral shunts. The hyperdynamic circulation is characterized by peripheral and splanchnic vasodilatation, reduced mean arterial pressure, and increased cardiac output. Vasodilatation, particularly in the splanchnic bed, permits an increase in inflow of systemic blood into the portal circulation.30
Splanchnic vasodilatation is caused in large part by relaxation of splanchnic arterioles and ensuing splanchnic hyperemia. Studies of experimental portal hypertension have demonstrated that splanchnic vascular endothelial cells are primarily responsible for mediating splanchnic vasodilatation and enhanced portal venous inflow through excess generation of NO.31–39 This excess generation of NO and ensuing vasodilatation, hyperdynamic circulation, and hyperemia in the splanchnic and systemic circulation contrasts with the hepatic circulation, in which NO deficiency contributes to increased intrahepatic resistance.
The mechanism of excess NO production from the endothelial cells of the systemic and splanchnic arterial circulation is an area of active investigation. Some of the increase in NO production probably occurs from shear stress–dependent and shear stress–independent increases in the expression of eNOS, which can be corrected in part by beta blockers.37,40–45 Activation of existing eNOS by cytokines or mechanical factors also seems to contribute to excess systemic and splanchnic NO generation through pathways that include eNOS phosphorylation and protein interactions.42–46 The physiologic stimuli that mediate this process are not well understood but may include ET-1, which is increased in the serum of patients with portal hypertension, and the cytokine tumor necrosis factor-α (TNF-α) because inhibitors of TNF improve portal pressure and the splanchnic circulatory disturbances in both human and experimental portal hypertension. TNF-α may be derived from intestinal endotoxin, and intestinal decontamination appears to correct the hyperdynamic circulation in humans, suggesting a link with intestinal inflammation.47 Vascular endothelial growth factor (VEGF) has also been implicated in this process by excessively activating eNOS.48 In humans with portal hypertension, therapeutic inhibition of NOS has met with mixed clinical results.
In one study, inhibition of NOS corrected altered systemic hemodynamics,49 but other studies have not demonstrated significant portal pressure–reducing effects of systemic NOS inhibition.50 Other mediators that may contribute to systemic and splanchnic vasodilatation include anandamide, an endogenous vasodilatory cannabinoid,51–53 heme oxygenase,17,54–56 and cyclooxygenase.57 Compelling evidence also supports a primary defect in smooth muscle cells in portal hypertension, perhaps because of defects in potassium channels.58–62 In fact, many pharmacologic therapies for portal hypertension target the splanchnic arteriolar smooth muscle cells, rather than endothelial cells, to reduce splanchnic vasodilatation. For example, octreotide, a synthetic analog of somatostatin, causes marked but transient reductions in portal pressure by contracting splanchnic smooth muscle cells, thereby limiting portal venous inflow, especially after meals. Nonselective beta blockers and vasopressin also reduce portal pressure by constricting splanchnic arterioles and thereby reducing portal venous inflow. Because intrahepatic resistance persists, therapies targeted toward the increase in portal venous inflow usually do not normalize portal pressure entirely but often blunt the prominent increases in portal venous inflow that occur in response to a meal. Combination therapy with an agent that reduces increased intrahepatic resistance, such as a nitrate, and an agent that reduces portal venous inflow, such as a beta blocker, are more effective in reducing portal pressure than is either agent alone.
COLLATERAL CIRCULATION AND VARICES
The portal vein–systemic collateral circulation develops and expands in response to elevation of the portal pressure.63 Blood flow in the low volumes that normally perfuse these collaterals and flow toward the portal circulation is reversed in portal hypertension because the increased portal pressure exceeds systemic venous pressure. Therefore, flow is reversed in these collateral vessels, and blood flows out of the portal circulation toward the systemic venous circulation.
Four distinct zones of venous drainage at the gastroesophageal junction are particularly relevant to the formation of esophageal varices.64 The gastric zone, which extends for 2 to 3 cm below the gastroesophageal junction, comprises veins that are longitudinal and located in the submucosa and lamina propria. They come together at the upper end of the cardia of the stomach and drain into short gastric and left gastric veins. The palisade zone extends 2 to 3 cm proximal to the gastric zone into the lower esophagus. Veins in this zone run longitudinally and in parallel in four groups corresponding to the esophageal mucosal folds. These veins anastomose with veins in the lamina propria. The perforating veins in the palisade zone do not communicate with extrinsic (periesophageal) veins in the distal esophagus. The palisade zone is the dominant watershed area between the portal and systemic circulations. More proximal to the palisade zone in the esophagus is the perforating zone, where there is a network of veins. These veins are less likely to be longitudinal and are termed perforating veins because they connect the veins in the esophageal submucosa and the external veins. The truncal zone, the longest zone, is approximately 10 cm in length, located proximally to the perforating zone in the esophagus, and usually characterized by four longitudinal veins in the lamina propria.
The fundus of the stomach drains through short gastric veins into the splenic vein. In the presence of portal hypertension, varices may therefore form in the fundus of the stomach. Splenic vein thrombosis usually results in isolated gastric fundal varices. Because of the proximity of the splenic vein to the renal vein, spontaneous splenorenal shunts may develop and are more common in patients with gastric varices than in those with esophageal varices.65,66
The predominant collateral flow pattern in intrahepatic portal hypertension is through the right and left coronary veins, with only a small portion of flow through the short gastric veins. Therefore, most patients with intrahepatic causes of portal hypertension have esophageal varices or gastric varices in continuity with esophageal varices. Unfortunately, portal hypertension caused by cirrhosis generally persists and progresses despite the development of even an extensive collateral circulation. Progression of portal hypertension results from (1) the prominent obstructive resistance in the liver; (2) resistance within the collaterals themselves; and (3) continued increase in portal vein inflow. The collateral circulatory bed develops through a combination of angiogenesis, the development of new blood vessels, and dilatation and increased flow through preexisting collaterals.3,67 Experimental evidence suggests that VEGF, a key NO stimulatory growth factor, may contribute to both the angiogenic and collateral vessel responses.55,68 Inhibition of VEGF or NO may attenuate the collateral vessel propagation by inhibiting angiogenic responses in experimental models of portal hypertension and collateralization.67–72 Some pharmacologic agents used in the management of portal hypertension, such as beta blockers and octreotide, may act in part by constricting collateral vessels.73–76 Approaches to inhibiting VEGF and angiogenesis are worth studying therapeutically.48
The development of gastroesophageal varices requires a portal pressure gradient of at least 10 mm Hg. Furthermore, a portal pressure gradient of at least 12 mm Hg is thought to be required for varices to bleed; other local factors that increase variceal wall tension also are needed77 because all patients with a portal pressure gradient of greater than 12 mm Hg do not necessarily bleed. Factors that influence variceal wall tension can be viewed in the context of the law of Laplace:
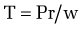
where T is variceal wall tension, P is the transmural pressure gradient between the variceal lumen and esophageal lumen, r is the variceal radius, and w is the variceal wall thickness. When the variceal wall thins and the varix increases in diameter and pressure, the tolerated wall tension is exceeded and the varix will rupture. These physiologic observations are manifested clinically by the observation that patients with larger varices (r) in sites of limited soft tissue support (w), with elevated portal pressure (P), tend to be at greatest risk for variceal rupture from variceal wall tension (T) that becomes excessive. One notable site in which soft tissue support is limited is at the gastroesophageal junction. The lack of tissue support and high vessel density may contribute to the greater frequency of bleeding from varices at the gastroesophageal junction. The law of Laplace also has implications for the relevance of pharmacologic therapies aimed at reducing portal pressure. Reductions in portal pressure will reduce the variceal transmural pressure gradient, thereby reducing the risk that variceal wall tension will become excessive and varices will rupture. Clinically, a reduction in the hepatic venous pressure gradient to less than 12 mm Hg almost negates the risk of variceal hemorrhage. The changes in portal pressure and local variceal factors, however, are dynamic and influenced by a number of physiologic (an increase in intra-abdominal pressure, meal-induced increases in portal pressure), diurnal (circadian changes in portal pressure), and pathophysiologic (acute alcohol use) factors, and portal pressure and esophageal variceal pressure may vary at different times.
MEASUREMENT OF PORTAL PRESSURE
HEPATIC VEIN PRESSURE GRADIENT
The HVPG is the difference between the wedged hepatic venous pressure (WHVP) and free hepatic vein pressure (FHVP). The HVPG has been used to assess portal hypertension since its first description in 1951,78 and has been validated as the best predictor for the development of complications of portal hypertension.
Measurement of the HVPG requires passage of a catheter into the hepatic vein under radiologic guidance until the catheter can be passed no further, that is, until the catheter has been “wedged” in the hepatic vein. The catheter can be passed into the hepatic vein through the femoral vein or using a transjugular venous approach. The purpose of wedging the catheter is to form a column of fluid that is continuous between the hepatic sinusoids and the catheter. Therefore, the measured pressure of fluid within the catheter reflects hepatic sinusoidal pressure. One of the drawbacks of using a catheter that is wedged in the hepatic vein is that the WHVP measured in a more fibrotic area of liver may be higher than the pressure measured in a less fibrotic area because of regional variation in the degree of fibrosis. Using a balloon-occluding catheter in the right hepatic vein to create a stagnant column of fluid in continuity with the hepatic sinusoids eliminates this variation in measurement of WHVP because the balloon catheter measures the WHVP averaged over a wide segment of the liver.79 HVPG is not effective for detecting presinusoidal causes of portal hypertension. For example, in portal hypertension secondary to portal vein thrombosis, the HVPG is normal. Moreover, the HVPG may underestimate sinusoidal pressure in primary biliary cirrhosis and other presinusoidal causes of portal hypertension.80 Therefore, HVPG is accurate for detecting only sinusoidal and postsinusoidal causes of portal hypertension.
The HVPG represents the gradient between the pressure in the portal vein and the intra-abdominal inferior vena caval pressure. An elevation in intra-abdominal pressure increases both WHVP and FHVP equally, so that the HVPG is unchanged. The advantage of the HVPG is that variations in the “zero” reference point have no impact on the HVPG.81 The HVPG is measured at least three times to demonstrate that the values are reproducible. Total occlusion of the hepatic vein by the inflated balloon to confirm that the balloon is in a wedged position is demonstrated by injecting contrast into the hepatic vein. A sinusoidal pattern should be seen, with no collateral circulation to other hepatic veins. The contrast washes out promptly with deflation of the balloon. Correct positioning of the balloon also is demonstrated by a sharp increase in the recorded pressure on inflation of the balloon. The pressure then becomes steady until the balloon is deflated, when the pressure drops sharply. In experienced hands, measurement of the HVPG is highly reproducible, accurate, and safe.
Measurement of the HVPG has been proposed for the following indications: (1) to monitor portal pressure in patients taking drugs used to prevent variceal bleeding; (2) as a prognostic marker82; (3) as an end-point in trials using pharmacologic agents for the treatment of portal hypertension83; (4) to assess the risk of hepatic resection in patients with cirrhosis; and (5) to delineate the cause of portal hypertension (i.e., presinusoidal, sinusoidal, or postsinusoidal (Table 90-1), usually in combination with venography, right-sided heart pressure measurements, and transjugular liver biopsy. Although the indication for HVPG measurement with the most potential for widespread use is monitoring the efficacy of therapies to reduce portal pressure, HVPG monitoring is not done routinely in clinical practice because no controlled trials have yet demonstrated its usefulness.84
PORTAL VEIN PRESSURE
Direct measurement of the pressure in the portal vein is a rarely used method that can be carried out through a percutaneous transhepatic route, transvenous approach, or, rarely, intraoperatively (although anesthesia can affect portal pressure). The transhepatic route requires portal vein puncture performed under ultrasound guidance. A catheter is then threaded over a guidewire into the main portal vein. With increasing use of the transjugular intrahepatic portosystemic shunt (TIPS) (see later), radiologists have gained expertise in puncturing the portal vein and measuring portal vein pressure by a transjugular route. Direct portal pressure measurements are carried out when HVPG cannot be measured, as in patients with occluded hepatic veins caused by the Budd-Chiari syndrome, in whom a surgical portosystemic shunt is being contemplated,85 or in patients with intrahepatic, presinusoidal causes of portal hypertension, such as idiopathic portal hypertension, in which the HVPG may be normal.
ENDOSCOPIC VARICEAL PRESSURE
Varices rupture and bleed when the expanding force of intravariceal pressure exceeds variceal wall tension. Measurement of the difference between intravariceal pressure and pressure within the esophageal lumen (the transmural pressure gradient across the varices) is potentially a more important indicator of bleeding risk than measurement of HVPG,86,87 especially in patients with portal vein thrombosis and other causes of portal hypertension associated with a normal HVPG.
A miniature pneumatic pressure sensitivity gauge attached to the tip of an endoscope (Varipres Solid Components, Barcelona, Spain) allows noninvasive measurement of variceal pressure. Patients with previous variceal bleeding have been demonstrated to have higher variceal pressures than those in patients without previous bleeding.88 A variceal pressure greater than 18 mm Hg during a bleeding episode is associated with failure to control bleeding and predicts early rebleeding.89 Moreover, patients on pharmacologic therapy who show a decrease in variceal pressure of greater than 20% from baseline have a low probability of bleeding, as compared with patients who do not demonstrate a greater than 20% decrease in variceal pressure, in whom the risk of variceal bleeding is 46%.88 Variceal pressure measurements determined with use of Varipres are considered satisfactory when they meet the following criteria: (1) a stable intraesophageal pressure; (2) absence of artifacts caused by esophageal peristalsis; and (3) correct placement of the gauge over the varix, as shown by fine fluctuations in the pressure tracing that correspond to the cardiac cycle and respirations. Therefore, measurement of variceal pressure requires both a skilled endoscopist and a cooperative patient, and, even in expert hands, accurate variceal pressure measurements cannot be obtained in 25% of patients.
Manometry using an endoscopic balloon to measure variceal pressure is subject to observer bias because it relies on visual appearance to determine whether the varices have collapsed.90–92 With this technique, a balloon is inserted into the esophagus and inflated until the varices are noted on endoscopy to collapse. The pressure in the balloon required to collapse the varices represents the variceal pressure. In general, techniques of measuring variceal pressure are still considered experimental and not suitable for routine clinical use.
DETECTION OF VARICES
UPPER GASTROINTESTINAL ENDOSCOPY
Upper gastrointestinal endoscopy is the most commonly used method to detect varices. The current consensus is that all patients with cirrhosis of the liver should be screened for esophageal varices by endoscopy. In patients in whom no varices are detected on initial endoscopy, endoscopy to look for varices should be repeated in 2 to 3 years. If small varices are detected on the initial endoscopy, endoscopy should be repeated in 1 to 2 years.93,94 None of the various noninvasive methods of determining which patients benefit most from endoscopic screening are accurate enough to recommend for routine use in clinical practice.95 The role of noninvasive markers in predicting the risk of large esophageal varices requires study in large multicenter trials. Preliminary data suggest that wireless video capsule endoscopy (see Chapter 19)96 and computed tomography (CT) imaging are alternative screening modalities in patients who are not candidates for upper endoscopy. Moreover, CT screening may be more cost-effective than endoscopy.97
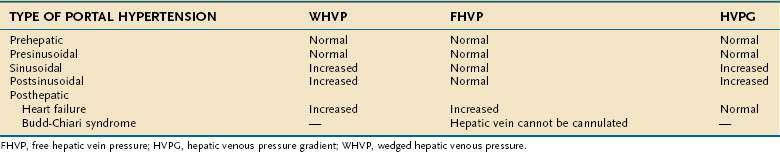
Endoscopic grading of esophageal varices is subjective. Various criteria have been used to try to standardize the reporting of esophageal varices. The best known of these criteria are those compiled by the Japanese Research Society for Portal Hypertension. The descriptors include red color signs, color of the varix, form (size) of the varix, and location of the varix.98 Red color signs include red “wale” markings, which are longitudinal whip-like marks on the varix; cherry-red spots, which usually are 2 to 3 mm or less in diameter; hematocystic spots, which are blood-filled blisters 4 mm or greater in diameter; and diffuse redness. The color of the varix can be white or blue. The form of the varix at endoscopy is described most commonly. Esophageal varices may be small and straight (grade I); tortuous and occupying less than one third of the esophageal lumen (grade II); or large and occupying more than one third of the esophageal lumen (grade III). Varices can be in the lower third, middle third, or upper third of the esophagus. Of all of the aforementioned descriptors, the size of the varices in the lower third of the esophagus is the most important. The size of the varices in the lower third of the esophagus is determined during withdrawal of the endoscope (Fig. 90-6). As much air as possible should be aspirated from the stomach while the esophageal lumen is fully inflated. Small varices, that is, those occupying less than one third of the lumen, are less than 5 mm in diameter, whereas large varices are greater than 5 mm in diameter.98,99 As a point of reference, any varix larger in diameter than an open pinch biopsy forceps is likely to be greater than 5 mm in diameter. Patients with large esophageal varices, Child (or Child-Pugh) class C cirrhosis (see later), and red color signs on varices have the highest risk of variceal bleeding within 1 year.100 The increase in bleeding risk attributable to the presence of red color signs, however, is not independent of the risk associated with large variceal size. Therefore, prophylactic treatment to prevent variceal bleeding is recommended in all patients with large esophageal varices irrespective of the presence or absence of red color signs (see later).
ULTRASONOGRAPHY
Ultrasound examination of the liver with Doppler study of the vessels has been used widely to assess patients with portal hypertension. Features suggestive of portal hypertension on ultrasonography include splenomegaly, portosystemic collateral vessels, and reversal of the direction of flow in the portal vein (hepatofugal flow). Some studies have demonstrated that a portal vein diameter greater than 13 mm and the absence of respiratory variations in the splenic and mesenteric veins are sensitive but nonspecific markers of portal hypertension.101,102 These criteria are not used routinely in clinical practice in most centers. Ultrasound examination can detect thrombosis of the portal vein, which appears as nonvisualization or cavernous transformation (a cavernoma) of the portal vein; the latter finding indicates an extensive collateral network in place of the portal vein.103 Splenic vein thrombosis also can be demonstrated. Portal blood flow can be measured by Doppler ultrasonography, which is the easiest research method for detecting postprandial increases in splanchnic blood flow.104 Although Doppler ultrasonography is clinically useful in the initial evaluation of portal hypertension, the technique is not widely used to provide quantitative assessments of the degree of portal hypertension. Transient elastography may be useful in detecting portal hypertension but is not sufficiently sensitive to recommend as a modality to monitor decreases in portal pressure in patients on pharmacotherapy (see Chapter 73).105
COMPUTED TOMOGRAPHY
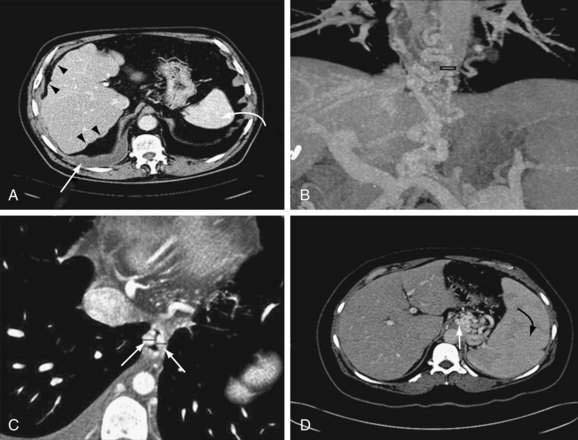
Computed tomography (CT) is useful for demonstrating many features of portal hypertension, including abnormal configuration of the liver, ascites, splenomegaly, and collateral vessels (Fig. 90-7). Detection of varices may be an emerging indication for CT. Diagnosis of fundal varices by multidetector row CT (MDCT) is at least as accurate as endoscopic ultrasonography (see later). CT is especially helpful in distinguishing submucosal from perigastric fundal varices106 and is considered a less invasive alternative to conventional angiographic portography in assessing portosystemic collaterals. At present, however, CT is not a recommended screening method for detecting large esophageal varices, but it may be a cost-effective method of screening for varices and preferred to endoscopy by patients.97
MAGNETIC RESONANCE IMAGING
Gadolinium-enhanced magnetic resonance imaging (MRI) is becoming recognized as a potentially useful method of detecting esophageal varices.107 In addition, MRI can be used to measure portal and azygous blood flow, which is increased in patients with portal hypertension.108 MRI provides excellent detail of the vascular structures of the liver and can detect portal venous thrombosis and spleen stiffness in patients with portal hypertension, but the role of MRI in the assessment of portal hypertension requires further study. Unlike transient elastography using ultrasound, MRI can accurately assess the stiffness of even fatty livers.109
ENDOSCOPIC ULTRASONOGRAPHY
Endoscopic ultrasound examination (endosonography) using radial or linear array echo-endoscopes or endoscopic ultrasound mini-probes passed through the working channel of a diagnostic endoscope has been applied as an investigational tool in the evaluation of patients with varices. Endoscopic ultrasonography has been used to study several aspects of esophageal varices, including the cross-sectional area of varices to identify patients at increased risk of bleeding77; size of and flow in the left gastric vein, azygous vein, and paraesophageal collaterals; changes after endoscopic therapy; and recurrence of esophageal varices following variceal ligation (see later).110 Endosonography can be combined with endoscopic measurement of transmural variceal pressure to allow estimation of variceal wall tension, which is a predictor of variceal bleeding (see earlier).111–113
CAUSES OF PORTAL HYPERTENSION
The usual classification of causes of portal hypertension is based on the site of increased resistance to portal blood flow—namely, prehepatic, intrahepatic, and posthepatic—and is outlined in Figure 90-5. Intrahepatic sites of increased resistance can be presinusoidal, sinusoidal, or postsinusoidal. Many causes of portal hypertension are associated with an increase in resistance at more than one site. For example, alcoholic cirrhosis may be associated with increased resistance at the presinusoidal, sinusoidal, and postsinusoidal levels. Therefore, classification based on the site of resistance may not be possible for all diseases that cause portal hypertension. A more useful classification is clinically based and considers common and less common causes of portal hypertension (Table 90-2).
Table 90-2 Causes of Portal Hypertension
COMMON CAUSES
Cirrhosis
Complications related to portal hypertension are the usual clinical manifestations of cirrhosis of the liver. Although all causes of cirrhosis are associated with portal hypertension, some features are disease specific. In alcoholic liver disease, elevation of the portal pressure is accurately reflected by the HVPG; moreover, portal hypertension may occur in the absence of cirrhosis but is more marked when cirrhosis is present. Perivenular lesions implicated in the pathogenesis in noncirrhotic alcoholic liver injury account for the presinusoidal component of portal hypertension in these patients.114 Autoimmune hepatitis also may be associated with portal hypertension in the absence of cirrhosis115; however, the risk of variceal bleeding is low in patients with autoimmune hepatitis. In patients with hemochromatosis, portal hypertension may be seen even before cirrhosis; the severity of portal hypertension increases with increasing fibrosis. Patients with hemochromatosis may bleed from varices despite an HVPG less than 12 mm Hg, indicating a presinusoidal component of portal hypertension. Phlebotomy therapy in patients with hemochromatosis may result in a decrease in portal hypertension.116 In patients with primary biliary cirrhosis, portal hypertension also may occur before cirrhosis has developed. The risk of variceal bleeding increases with an increase in the histologic stage of the disease.117 In earlier stages of primary biliary cirrhosis, portal hypertension is predominantly presinusoidal, but as the disease progresses, a sinusoidal component develops. Therefore, the HVPG may underestimate portal pressure in patients with primary biliary cirrhosis.80 Portal hypertension occurs in patients with primary sclerosing cholangitis and in those with biliary strictures. A long duration of biliary obstruction usually is required, although portal hypertension has been known to develop in a few months in patients with chronic bile duct obstruction caused by chronic alcoholic pancreatitis.118 Portal hypertension in patients with biliary obstruction regresses following relief of the biliary obstruction.
Schistosomiasis
Schistosomiasis may be the most common cause of portal hypertension worldwide (see Chapter 82). Bleeding from esophageal varices is a major cause of death in patients with hepatosplenic schistosomiasis. Portal hypertension results from presinusoidal obstruction caused by deposition of eggs of Schistosoma mansoni and Schistosoma japonicum in the presinusoidal portal venules. The host reaction results in granulomatous inflammation, which causes presinusoidal and periportal fibrosis.119 The fibrosis that results is sometimes called “clay pipestem” or simply “pipestem” fibrosis and usually is associated with sustained heavy infection. The periportal collagen deposition leads to progressive obstruction of portal blood flow, portal hypertension, and variceal bleeding, along with splenomegaly and hypersplenism. Lobular architecture usually is preserved. Coinfection with hepatitis B or C virus in patients with hepatic schistosomiasis can result in more rapid progression of fibrosis, hepatic failure, and an increased risk of hepatocellular carcinoma.120
Extrahepatic Portal Vein Thrombosis
Extrahepatic portal vein thrombosis is a prehepatic, presinusoidal cause of portal hypertension and a common cause of portal hypertension in children (see Chapter 83). The most common causes of portal vein thrombosis include hematologic disorders such as polycythemia vera or other myeloproliferative disorders. Other causes include a prothrombotic state, such as antithrombin, protein C, or protein S deficiency; antiphospholipid syndrome (or antiphospholipid antibody syndrome); paroxysmal nocturnal hemoglobinuria; oral contraceptive use; a neoplasm, usually intra-abdominal; an inflammatory disease, such as pancreatitis, inflammatory bowel disease, or diverticulitis; abdominal trauma; and postoperative states, especially postsplenectomy. Cirrhosis is a cause of portal vein thrombosis.121 Older studies suggested that portal vein thrombosis occurs in approximately 6% of patients with cirrhosis and in up to 25% of those with cirrhosis and hepatocellular carcinoma.122 With improved imaging, portal vein thrombosis is now known to be a more common complication of cirrhosis, and the association with hepatocellular carcinoma may not be as strong as previously thought. Isolated splenic vein thrombosis caused by a pancreatic neoplasm or pancreatitis usually is not associated with a thrombophilia. Umbilical vein sepsis may be an etiologic factor in children with portal vein thrombosis, but even in these cases, an associated prothrombotic state may predispose the patient to portal vein thrombosis.
Acute and subacute portal vein thrombosis usually does not manifest with variceal bleeding.1 Chronic portal vein thrombosis is suggested by nonvisualization of the portal or splenic vein and an extensive collateral circulation. Patients may present with nonspecific symptoms or with variceal bleeding and hypersplenism. Bleeding usually is from gastroesophageal varices but may be from duodenal varices and, rarely, other ectopic sites. Gallbladder varices (Fig. 90-8) also have been described in patients with portal vein thrombosis.123
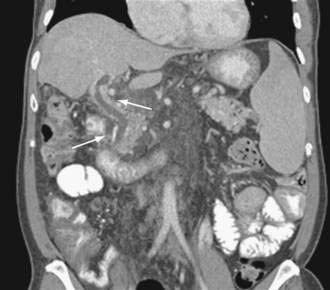
The treatment of portal vein thrombosis is symptomatic, with the aim of controlling variceal bleeding or preventing recurrent variceal bleeding. Patients in whom esophageal varices are not large, and a thrombophilia is detected, are best managed with anticoagulation because in these patients, the benefits of anticoagulation outweigh the risks.124 Local or systemic thrombolytic therapy is seldom required and is generally reserved for patients in whom a portal vein thrombus extends into the superior mesenteric vein, with danger of impending intestinal ischemia. Endoscopic therapy is used to control acute variceal bleeding and to prevent recurrent bleeding. Use of pharmacologic agents such as beta blockers to prevent variceal bleeding is probably also effective in patients with portal vein thrombosis, but this approach has not been well studied. Patients with portal vein thrombosis have lower mortality and morbidity rates from variceal bleeding than those reported in patients with cirrhosis and variceal bleeding, owing to the lack of coagulopathy and synthetic liver dysfunction. Surgical portosystemic shunt procedures are carried out in patients in whom bleeding cannot be controlled by conservative measures. If a suitable vein is not available for anastomosis, a large collateral vein may be anastomosed to a systemic vein.125 Placement of a TIPS is possible in some patients with chronic portal vein thrombosis.
Idiopathic Portal Hypertension
Idiopathic portal hypertension is uncommon in Western countries but is common in parts of Asia such as India and Japan. This disorder is diagnosed when the portal pressure is elevated in the absence of significant histologic changes in the liver or extrahepatic portal vein obstruction.126 A liver biopsy specimen from affected patients may be entirely normal,122 although increased concentrations of ET-1 have been noted in the periportal hepatocytes, portal venules, and hepatic sinusoids of patients with idiopathic portal hypertension.127 Various terms used (rather loosely) to describe idiopathic portal hypertension include hepatoportal sclerosis, noncirrhotic portal fibrosis, and Banti’s syndrome.128,129 Use of the term idiopathic portal hypertension probably is best restricted to portal hypertension in patients in whom no hepatic lesion is found on light microscopy. The term hepatoportal sclerosis suggests obliterative portal venopathy with subendothelial thickening of the intrahepatic portal veins; thrombosis and recanalization of these veins may follow. Fibrosis of the portal tracts is prominent later in the course.
The cause of idiopathic portal hypertension is unclear in a majority of patients, although chronic arsenic intoxication, exposure to vinyl chloride, and hypervitaminosis A have been implicated (see Chapter 87). These etiologic factors are present in only a minority of patients. The dominant clinical features of the condition are variceal bleeding and hypersplenism related to a markedly enlarged spleen. Liver biochemical test levels are usually normal, although the serum alkaline phosphatase level may be mildly elevated. Ascites is uncommon. The HVPG in this disorder usually is normal because the site of increased resistance is presinusoidal.130 Surgical portosystemic shunts are well tolerated in these patients, although hepatic encephalopathy may occur on long-term follow-up evaluation.122 Liver transplantation is rarely required in these patients.
Idiopathic portal hypertension may be confused with incomplete septal cirrhosis, which probably is an unrelated condition characterized by incomplete septa and liver nodularity.131 Patients with incomplete septal cirrhosis are clinically similar to patients with cirrhosis and may progress to end-stage liver disease and require liver transplantation.
LESS COMMON CAUSES
Nodular Regenerative Hyperplasia
Nodular regenerative hyperplasia is a histopathologic diagnosis characterized by atrophy of zone 3 hepatocytes and hypertrophy of zone 1 hepatocytes without significant fibrosis (see Chapters 35 and 94).132 This disorder has been recognized increasingly as a cause of portal hypertension and may even occur after liver transplantation.133 Similar histologic changes may be seen in well-established Budd-Chiari syndrome.134 The nodular hyperplasia may not be apparent on histologic examination unless a reticulin stain is carried out to demonstrate the micronodules. These regenerative nodules are believed to result from an imbalance between hyperperfused areas of the liver, with resulting regenerative nodules, and poorly perfused areas, with resulting atrophy. Nodular regenerative hyperplasia is associated with a variety of conditions, predominantly hematologic and rheumatologic in nature. Liver biochemical abnormalities include mild elevation of the serum aminotransferase levels. Portal hypertension manifesting as variceal bleeding is the predominant clinical presentation. Ascites also may develop in these patients, suggesting that an increase in sinusoidal pressure occurs.135 Hepatocellular carcinoma does not occur, but liver transplantation may be required in some patients.
Partial Nodular Transformation of the Liver
Partial nodular transformation of the liver is an uncommon lesion that is characterized by large nodules in the perihilar region.136 These nodules may be visible on imaging studies of the liver. The rest of the liver may be normal or may show changes of nodular regenerative hyperplasia. Liver biochemical test levels usually are normal. Like nodular regenerative hyperplasia, partial nodular transformation of the liver is believed to be related to an imbalance in portal perfusion of the liver, but the abnormality is restricted to the hilar branches, whereas in nodular regenerative hyperplasia the abnormality is more diffuse. Variceal bleeding is the predominant presentation in partial nodular transformation of the liver, although patients with large nodules may experience abdominal pain. Hepatocellular carcinoma may rarely develop in these regenerating nodules. Treatment with a surgical portosystemic shunt is associated with good long-term results.
Fibropolycystic Liver Disease
Fibropolycystic liver disease is a term which encompasses Caroli’s disease, Caroli’s complex (Caroli’s disease with congenital hepatic fibrosis), congenital hepatic fibrosis, and polycystic liver disease. Congenital hepatic fibrosis usually occurs in association with Caroli’s disease of the liver, polycystic disease of the kidney, and medullary sponge kidney (see Chapter 62). The major manifestation of congenital hepatic fibrosis is variceal bleeding.137
Related posts:
 Surgical Peritonitis and Other Diseases of the Peritoneum, Mesentery, Omentum, and Diaphragm
Surgical Peritonitis and Other Diseases of the Peritoneum, Mesentery, Omentum, and Diaphragm
 Hepatitis Caused by Other Viruses
Hepatitis Caused by Other Viruses
 Hemochromatosis
Hemochromatosis
 Gastrointestinal and Hepatic Complications of Solid Organ and Hematopoietic Cell Transplantation
Gastrointestinal and Hepatic Complications of Solid Organ and Hematopoietic Cell Transplantation
 Nutritional Assessment and Management of the Malnourished Patient
Nutritional Assessment and Management of the Malnourished Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


