Fig. 4.1
Adipocyte dysfunction in obesity . Adipocytes hypertrophy and enlarge during obesity. This can lead to an imbalance between blood flow and oxygen demand, resulting in hypoxia and cell death. Dysfunctional adipocytes produce pro-inflammatory cytokines including TNF and CCL2. TNF induces insulin resistance, which leads to lipid hydrolysis and release of fatty acids into the circulation. CCL2 recruits macrophages, which produce more cytokines and amplify the inflammatory and insulin-resistant state
Epidemiologic studies suggest that certain adipose tissue depots have a greater potential for dysfunction in obesity than others. Specifically, visceral fat is highly prone to insulin resistance [17], and visceral obesity often correlates with hepatic steatosis or liver injury [18–22]. Interestingly, though, the connection between visceral obesity and hepatic steatosis in humans may not be a direct consequence of insulin resistance. This was highlighted in the Dallas Heart Study, in which African-Americans with visceral obesity exhibited more insulin resistance than Hispanics but had less hepatic steatosis [21].
Although hepatic steatosis typically follows obesity, one important exception to this rule is that hepatic steatosis and even steatohepatitis also occur in patients with lipodystrophy, characterized by a paucity of adipose tissue [23–25]. This paradox can be explained by the fact that normal adipose tissue produces leptin, adiponectin, and other adipokines whose functions are to promote lipid homeostasis [23, 26]. In the setting of lipodystrophy, the absence of functional adipose tissue induces the same adverse consequences for the liver as does the presence of dysfunctional adipose tissue in obesity. When viewed together, both diseases underscore that some amount of healthy adipose tissue is required to maintain metabolic homeostasis in the liver [27].
Adipose Tissue Insulin Resistance and Downstream Consequences for the Liver
As mentioned above, insulin-resistant adipose tissue loses its ability to store lipid and releases free fatty acids into the circulation. In an effort to compensate for adipose tissue insulin resistance, the pancreas produces more insulin, which leads to hyperinsulinemia . Thus, in obesity, the liver encounters excesses of both free fatty acids and insulin (Fig. 4.2a). Liver cells take up fatty acids through the transport proteins FATP5 (fatty acid transport protein 5) and CD36 [28, 29]. Both proteins are upregulated in obesity [30, 29], priming the liver to accommodate the enhanced flux from adipose tissue. Once inside the liver, fatty acids are steered toward storage in the form of triglyceride, which provides an immediate and direct route to hepatic steatosis. During the process of triglyceride synthesis, the levels of several intermediates in the triglyceride pathway also rise. Of particular importance are diacylglycerols (DAG), which can activate protein kinase C epsilon (PKCε). PKCε, in turn, can interfere with the normal function of insulin receptors on hepatocytes [31, 32], resulting in a state of hepatic insulin resistance.
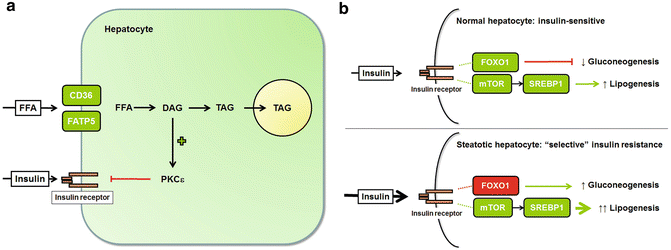
Fig. 4.2
Hepatic steatosis promotes hepatic insulin resistance. (a) Insulin resistance outside the liver exposes the liver to high circulating levels of free fatty acids (FFA) and insulin. FFA are taken up by hepatocytes via the transporters CD36 and FATP5. Once inside the cell, the FFA are incorporated into diacylglycerols (DAG) and triacylglycerols (TAG). Excess DAG within hepatocytes activates PKCε, which impairs insulin signaling. (b) In normal hepatocytes, insulin stimulates lipogenesis and suppresses gluconeogenesis. Lipogenesis is upregulated by mTOR and SREBP1; gluconeogenesis is suppressed by FOXO1. In obesity, circulating insulin levels are high, and hepatocyte insulin signaling is impaired, at least partially. Insulin-mediated FOXO1 activation is impaired, resulting in uncontrolled gluconeogenesis; insulin-mediated activation of mTOR and SREBP1, however, is not suppressed, which leads to enhanced lipogenesis. These discordant responses have been termed “selective insulin resistance”
In the liver, insulin is an important regulator of two metabolic functions: gluconeogenesis and lipid synthesis. Gluconeogenesis is normally suppressed by insulin, and thus, hepatic insulin resistance is marked by an increase in hepatic glucose production (Fig. 4.2b). Hepatic lipid synthesis is normally enhanced by insulin; thus, hepatic lipid synthesis should be decreased in an insulin-resistant liver. Curiously, this is not the case, and the paradox has been referred to as “selective insulin resistance ” (Fig. 4.2b) [33]. The fact that hepatic lipid synthesis remains sensitive to insulin in a state of hepatic lipid excess whereas gluconeogenesis does not implies that the two metabolic pathways must diverge at some point. Experimental studies indicate that gluconeogenesis is controlled by forkhead box O1 (FOXO1) downstream of the insulin receptor, whereas lipogenesis is controlled by mammalian target of rapamycin (mTOR) [34]. This leaves open the possibility that in a state of hepatic insulin resistance, other factors besides insulin could continue to stimulate hepatic lipogenesis by activating mTOR.
The lipogenic program activated in the liver during obesity involves more than the esterification of adipocyte-derived fatty acids to triglyceride. It also involves the synthesis of new fatty acids from carbohydrate (de novo lipogenesis; DNL) (Fig. 4.3). The enzymes responsible for DNL are regulated in part by insulin, through the action of the transcription factor sterol regulatory element-binding protein-1 (SREBP1) [35]. Importantly, SREBP1 is active in the liver even during times of hepatic insulin resistance (see above, “selective insulin resistance”). DNL enzymes are also regulated by glucose through the action of a second transcription factor, carbohydrate response element-binding protein (ChREBP) . Together, SREBP1 and ChREBP promote hepatic DNL in response to high-carbohydrate feeding, hyperglycemia, and hyperinsulinemia [35]. In the normal liver, only about 4 % of hepatic triglyceride derives from DNL. In obese individuals, this proportion increases as fatty liver progresses, to as much as 25 % [36, 37]. Because the flux of fatty acids to the liver from adipose tissue does not decline as DNL rises [38, 39], adipose tissue-derived fatty acids must over time be diverted to pathways other than triglyceride synthesis. Evidence indicates they are shuttled to oxidation, to provide the energy necessary to drive the processes of gluconeogenesis and DNL [39].
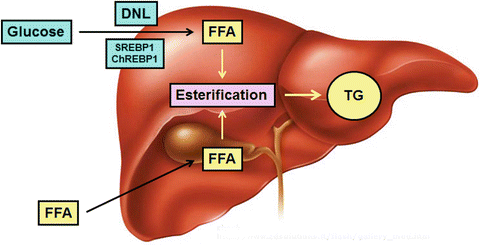
Fig. 4.3
Hepatic steatosis arises from de novo lipogenesis as well as fatty acid uptake. Excess free fatty acids (FFA) in the liver are esterified to triglyceride (TG) for storage. Some FFA are taken up by the liver from the circulation; others are produced in the liver from glucose, through de novo lipogenesis (DNL). DNL is stimulated by insulin (via SREBP1) and glucose (via ChREBP1)
ER Stress and Its Contribution to Hepatic Steatosis
Once lipid accumulation begins in hepatocytes, it can set into motion events that prompt even more steatosis. A key contributor to this process is the endoplasmic reticulum (ER), an organelle that plays a central role in cellular metabolism. The classical function of the ER is to execute posttranslational processing of proteins. The ER can become stressed, either from an increased demand for protein folding or from derangements in the internal environment of the organelle [40]. Stress in the ER prompts adaptive responses involving the activation of one or more ER membrane proteins as shown in Fig. 4.4. Although these responses are designed to maintain ER homeostasis, they may have undesirable side effects. In hepatocytes, for example, exposure to excess fatty acids places stress on the ER by creating demand for the synthesis of apolipoproteins needed for lipid export as VLDL [18]. This activates the RNA-dependent protein kinase-like ER kinase (PERK), which signals downstream events that result in a suppression of cellular protein synthesis. Although well intentioned, this response prevents rather than facilitates fatty acid export; the ensuing outcome is fatty acid retention and steatosis.
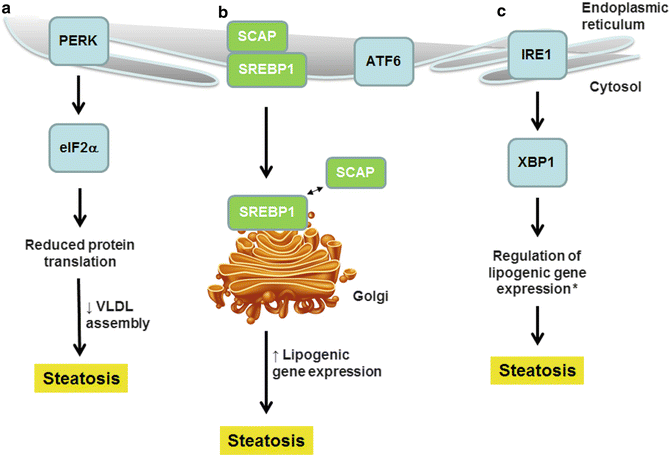
Fig. 4.4
Endoplasmic reticulum stress contributes to hepatic steatosis. ER stress transducers, positioned within the ER membrane, respond to excess fatty acids. (a) Activation of PERK leads to a reduction in protein synthesis, including the synthesis of apolipoproteins necessary for VLDL assembly. (b) SREBP1, although not itself an ER stress transducer, is activated by ER stress similarly to ATF6. ER stress promotes migration of SREBP1 to the Golgi where it becomes activated by dissociation from SCAP; SREBP1 stimulates lipogenic gene expression. (c) Activation of IRE1 leads to activation of XBP1, a transcription factor that regulates lipogenic gene expression. These three events promote hepatic steatosis (*whether XBP1 stimulates or suppresses lipogenic gene expression is currently debated)
Another important consequence of ER stress is the transformation of SREBP1 from an inactive to a transcriptionally active form [41]. SREBP1, like the ER stress proteins shown in Fig. 4.4, normally resides in the ER membrane; there it is maintained in an inactive state through interactions with a binding partner named SCAP (SREBP cleavage-activating protein) [42]. Under conditions of ER stress, the SREBP1–SCAP complex moves from the ER to the Golgi apparatus; the complex then dissociates, and SREBP1 is released from the Golgi membrane by proteolytic cleavage, enabling its translocation to the nucleus [43]. A connection between ER stress and SREBP1 activation was first theorized when it was noted that the same steps of Golgi transport and membrane release required to process SREBP1 are also needed to activate the ER stress transducer ATF6 (activating transcription factor 6) [44]. Subsequently, experiments showed that ER stress can activate SREBP1 independently of its classic stimulus, insulin. This was a key finding because it revealed yet another potential mechanism by which SREBP1 and hepatic lipogenesis could persist in fatty livers in the face of hepatic insulin resistance. Importantly, if hepatic steatosis induces ER stress in the liver, and ER stress in turn activates SREBP1, this creates a positive feedback loop for the persistence and progression of fat accumulation in the livers of obese individuals once the process is initiated.
Among the adaptive responses to ER stress is an increase in the synthesis of cellular lipids. Lipid synthesis enables a stressed ER to expand its volume and processing capacity [45]. Stress-related lipid synthesis is mediated primarily through inositol-requiring enzyme 1 (IRE1) and its downstream target X-box protein-1 (XBP1) [46–48]. These two molecules have the potential to influence several aspects of lipid metabolism, including fatty acid and triglyceride synthesis, lipid hydrolysis, and lipid transport [49, 50]. In the liver, the lipid alterations that ensue from activation of IRE1 and XBP1 may not always be adaptive. Instead, they may contribute to hepatic steatosis. This was first discovered when disruption of the pathway by silencing XBP1 in the livers of obese mice improved hepatic steatosis [51]. Recent research into the role of the IRE1–XBP1 axis in hepatic lipid homeostasis has cast some doubt on the ability of IRE1 and XBP1 to stimulate lipogenesis directly. In fact, one group suggests that IRE1–XBP1 signaling is actually necessary for the successful lipidation of VLDL particles [49]. In this case, activation of IRE1–XBP1 would prevent, rather than promote, hepatic steatosis.
Although at present there remains some uncertainty about the importance of ER stress in comparison to other mechanisms of obesity-related hepatic steatosis (Table 4.1), there is ample experimental evidence that ER stress regulates hepatic lipid homeostasis at multiple levels through multiple signal transducers. ER stress signaling can also lead to hepatocyte death in a fatty liver; this will be discussed below.
Table 4.1
Factors leading to hepatic steatosis in obesity
Factor | Mechanism |
|---|---|
Increased flux of FFA from the adipose tissue to liver | Insulin resistance in adipose tissue promotes lipolysis and FFA release |
Increased hepatic uptake of FFA from the circulation | Upregulation of hepatic fatty acid transporters CD36 and FATP5 |
Insulin-mediated stimulation of hepatic lipogenesis | Insulin-dependent stimulation of the lipogenic transcription factor SREBP1 |
Glucose-mediated stimulation of hepatic lipogenesis | Glucose-dependent stimulation of the lipogenic transcription factor ChREBP1 |
ER stress-mediated stimulation of hepatic lipogenesis | Insulin-independent activation of SREBP1; IRE1–XBP1 activation leading to induction of lipogenic genes |
ER stress-mediated inhibition of hepatic lipid export | PERK-mediated reduction of ApoB synthesis, resulting in reduced VLDL synthesis |
Autophagy and Steatosis
Autophagy is a process that degrades dysfunctional cellular constituents and maintains cellular energy stores. During autophagy, material is captured in a membrane-bound vesicle called an autophagosome, which then fuses with a lysosome permitting digestion of the contents and releases back into the cytosol. Recently, it was discovered that autophagy plays an important role in lipid homeostasis in adipocytes [52] and hepatocytes [53]. Autophagy is considered particularly important to hepatocytes as a means of accessing the energy stored in cellular lipid droplets, because these cells are not equipped with high concentrations of lipases [54]. Fatty acids mobilized from lipid droplets via autophagy are routed to mitochondria for oxidation. The importance of autophagy to normal hepatic lipid metabolism was documented in experiments demonstrating that inhibition of autophagy leads to spontaneous hepatic steatosis [53]. More importantly, and directly pertinent to fatty liver disease, it was discovered that loading normal liver cells with lipid impairs autophagy [53]. This prompted further investigation to determine whether autophagy is dysregulated in a fatty liver in vivo, and the suspicion was confirmed [53]. At present, it does not appear that abnormal autophagy is a primary cause of hepatic steatosis, but impairments in autophagy that result from steatosis can enhance or perpetuate hepatic lipid accumulation. Dysregulated autophagy can also promote cell death in a fatty liver; this is discussed later (see section “Mechanisms of Cell Death in NASH”).
Regulation of Steatosis by Bile Acids
Bile acids exert important influences on lipid metabolism in the liver [55]. The principal means by which bile acids control lipid homeostasis is through interactions with the farnesoid X receptor (FXR) (Fig. 4.5). Bile acid-induced stimulation of FXR leads to activation of the downstream target, short heterodimer partner (SHP); SHP, in turn, suppresses SREBP1, which results in inhibition of hepatic lipogenesis. FXR also activates PPARα, a master inducer of fatty acid oxidation. This means that bile acid signaling should limit hepatic steatosis by two complementary means. The tonic influence of bile acids on hepatic lipid homeostasis has been demonstrated in mouse studies, which reveal that knockout of FXR leads to hepatic steatosis and steatohepatitis [56]. On the other hand, there is only limited evidence that an alteration in FXR is a root cause of fatty liver disease in humans. One study reports that patients with hepatic steatosis and NASH have low levels of FXR in the liver [57]. Another looked for modulation of hepatic lipid levels as a consequence of a polymorphism in the gene encoding FXR, but found none [58]. Recently, however, it was discovered that high-fat feeding in mice induces a gene named yin yang 1 (YY1) that suppresses FXR [59]. YY1 is upregulated by TNF [60], and thus, the pro-inflammatory state of obesity may be responsible for YY1 induction and FXR suppression.
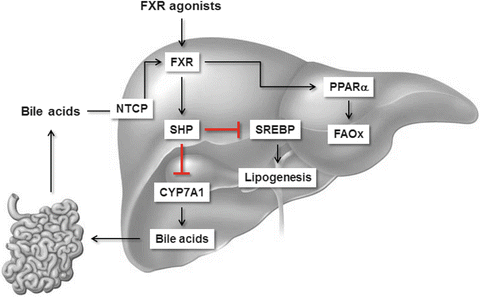
Fig. 4.5
Regulation of hepatic lipid metabolism by bile acids. Bile acids entering the liver via the NTCP transporter stimulate the nuclear hormone receptor, FXR. FXR then activates SHP, which suppresses bile acid synthesis and also suppresses hepatic lipogenesis by reducing SREBP1 activity. FXR also activates PPARα, which stimulates fatty acid oxidation (FAOx). Taken together, FXR activation suppresses hepatic lipid synthesis and enhances fatty acid oxidation. Pharmacologic FXR agonists have similar beneficial effects on hepatic lipid metabolism
Influence of Genetics on Hepatic Steatosis
There is a heritable element to NAFLD and NASH, based on evidence that the disease clusters in families, is concordant among monozygotic twins, and exhibits clear ethnic variations in disease risk (reviewed in [65]). The genetics of NAFLD and NASH are discussed in detail in this chapter. Because the current chapter focuses on the pathogenesis of NAFLD and NASH, it is worth highlighting a few examples of NASH-related gene polymorphisms here, focusing on the mechanism by which they might contribute to disease pathogenesis. The most notable genetic alteration that has been linked to NASH is a C → G polymorphism leading to an I148M substitution in the gene PNPLA3 (patatin-like phospholipase domain-containing 3) [66–68]. PNPLA3 encodes adiponutrin, a protein located in the membrane fraction of hepatocytes and adipocytes. Investigators have gone to great lengths to elucidate how mutant adiponutrin promotes steatosis. In one study, mice were engineered to overexpress human I148M adiponutrin. They exhibited enhanced hepatic lipogenesis, which led to the conclusion that adiponutrin functions primarily as lysophosphatidic acid acyltransferase (LPAAT) that generates lysophosphatidic acid in the pathway leading to triacylglycerols [69]. However, in a more recent study, the I148M mutation was “knocked into” the native mouse gene. Using this approach, the mice displayed impaired triglyceride lipolysis, prompting the opposite conclusion that adiponutrin is primarily a triglyceride lipase [70]. Although more research is required to clarify the precise role of mutant adiponutrin in fatty liver disease, some observations are worth noting. First, the I148M knock-in mutation does not cause spontaneous hepatic steatosis in mice, but does enhance steatosis in response to high-carbohydrate feeding [70]. This is consistent with the classification of PNPLA3 as a modifier of NASH in humans [71]. Second, although overexpression or knock-in of mutant adiponutrin induces hepatic steatosis in mice, as yet there is no report of its ability to induce experimental steatohepatitis.
A gene named transmembrane 6 superfamily member 2 (TM6SF2) was recently discovered in an exome-wide association study to be associated with hepatic steatosis [72]. The link between TM6SF2 and NAFLD is believed to supersede that of a nearby gene, neurocan (NCAN), which was uncovered in an earlier GWAS study [67]. Hepatic steatosis is associated with an A → G polymorphism in TM6SF2 leading to a glutamate-to-lysine substitution at amino acid 167. The TM6SF2 polymorphism has already been confirmed by other groups as relevant not only to steatosis [73] but also NASH and cirrhosis [74]. TM6SF2 is a transmembrane protein of unknown function, but in vitro studies indicate the E167K variant has reduced stability. Knockdown of TM6SF2 in liver cell lines and mouse liver in vivo results in steatosis, particularly following a carbohydrate challenge. Importantly, TM6SF2-deficient mice have reduced levels of circulating lipids, which suggests the protein facilitates hepatic VLDL secretion [72]. These examples illustrate how individual gene polymorphisms, each with a strong association to NAFLD but an independent effect on the liver, can impact the NAFLD phenotype. They also underscore how multiple independent modifiers in an individual subject might complicate disease severity and response to treatment.
Influence of the Gut Microbiome on Hepatic Steatosis
It is now evident that microbial communities in the intestine are important determinants of metabolism. Pertinent to obesity are observations that diet affects the gut microbiome and the gut microbiome in turn affects nutrient absorption. Specifically, overeating stimulates the intestinal overgrowth of bacteria belonging to the phylum Firmicutes [75, 76], which are efficient at extracting nutrients from the diet [77]. Some of the adverse effects of round-the-clock eating may also stem from alterations in the gut microbiome [78]. Thus, microbial derangements induced by alterations in eating behavior create a situation that enhances the potential for obesity and its complications.
In addition to having an altered colonic microbiome, overweight individuals with fatty livers often display small intestinal bacterial overgrowth (SIBO) [79, 80]. SIBO is thought to arise from impaired intestinal motility in obesity, which can result from a reduction in the hormone ghrelin [81]. Toxins or inflammatory mediators elaborated by these abundant organisms can compromise intestinal barrier function by degrading tight junction proteins [80, 82]. This allows potentially harmful compounds from the intestine to invade the portal circulation and reach the liver, where they can stimulate NAFLD and NASH.
Two intestinal compounds that have been implicated in the development of NAFLD and NASH are bacterial endotoxin and ethanol. Endotoxin , which can enter the portal circulation through a compromised intestinal barrier, is readily detectable in the blood of patients with hepatic steatosis and NASH [80, 83]. Importantly, endotoxin has even been identified in the blood of healthy persons fed a high-energy diet for a brief period [82, 84]. This suggests that alterations in the intestine can occur very early in the evolution of obesity and NAFLD. Ethanol, a by-product of bacterial fermentation in the gut, can be absorbed through an intact intestinal epithelium. Like endotoxin, ethanol is detectable in the circulation of patients with NAFLD [85, 86] and has the potential to promote hepatic steatosis or steatohepatitis (see Chap. 9A on Alcoholic Liver Disease).
Intestinal bacteria can also perform other functions that lead to undesirable effects on hepatic lipid homeostasis. One is the ability to convert choline into methylamines. This property, which is characteristic of microbes that overgrow in obesity [87], can lead to choline depletion in the intestine and choline deficiency in the host. Choline deficiency is well known to promote hepatic steatosis [88]; it does so by limiting the synthesis of phosphatidylcholine, critical to the assembly of VLDL particles [89]. Intestinal bacteria also suppress the synthesis of fasting-induced adipocyte factor (FIAF), an inhibitor of lipoprotein lipase [90, 91]. When FIAF is reduced, adipose tissue lipolysis is increased, which results in an increase in fatty acid delivery to the liver.
Lastly, gut microbes can influence metabolism through effects on bile acids. Gut bacteria can modify bile acid structure and activity; conversely, bile acids can influence microbial homeostasis in the gut. The interplay between microbes and bile acids, as well as other topics relevant to the gut microbiome in fatty liver disease, is reviewed in [92–94].
Behavioral Factors Affecting Hepatic Steatosis
Intake of Specific Dietary Nutrients
Although there is an undeniable relationship between caloric intake and obesity/fatty liver disease, studies suggest not all calories are alike. Indeed, certain nutrients have a specific propensity to promote steatosis and steatohepatitis. Among the dietary sugars, fructose is considered particularly noxious because it induces visceral obesity and insulin resistance more readily than glucose [95]. This is likely attributable to the unregulated nature of fructose entry into DNL, as well as the ability of fructose to directly activate SREBP1 independently of insulin [96, 97]. Epidemiologic studies implicate fructose intake as a risk factor for NAFLD in adults and children [98–100]. Moreover, adding fructose to a standard diet in the form of sweetened beverages can provoke hepatic steatosis in as few as 7 days [101]. Although there remains some controversy whether fructose is more harmful to the liver than other sugars [102], head-to-head comparisons in animal models support this theory [103].
Of the dietary fats, trans fats, which are man-made unsaturated fats, have a specific tendency to provoke NAFLD. Data supporting the connection between trans fats and NAFLD come largely from animal studies. In mice, trans fats produce more steatosis and liver injury than saturated or standard polyunsaturated fats when consumed in isocaloric amounts [104, 105]. Interestingly saturated fats, which are singled out as important risk factors for cardiovascular disease [106], do not consistently induce more hepatic steatosis than unsaturated fats in mice when incorporated into experimental diets [107, 108]. When saturated fats are paired with simple sugars, though, the two nutrients synergize to promote hepatic steatosis (Maher JJ et al., unpublished). This may be due to the ability of saturated fat to stimulate hepatic DNL by activating SREBP1 through peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1α) [109]. In humans, saturated fat-rich food supplements promote more hepatic steatosis than polyunsaturated fat-rich supplements [110]. Overall, the available information to date indicates that saturated fats and trans fats both have greater potential than other types of fat to induce hepatic steatosis and liver injury.
Cholesterol consumption, which has been linked to the development of chronic liver disease in humans [111], contributes directly to the pathogenesis of experimental NASH. Mice fed diets containing 0.2–2.0 % cholesterol by weight consistently develop more severe steatohepatitis than those fed diets lacking cholesterol [112–116]. Fatty liver disease in cholesterol-fed mice coincides with the accumulation of free cholesterol in the liver [115], which may contribute directly to liver cell injury (see section “Mechanisms of Cell Death”). A similar rise in hepatic free cholesterol has been reported in humans with NAFLD and NASH [117]. The fact that cholesterol is elevated in NAFLD as well as NASH could indicate this derangement is a prerequisite to disease progression.
The Diurnal Clock and the Timing of Food Intake
Many bodily functions are subject to diurnal control through the action of the so-called clock genes. A 24-h oscillatory cycle of gene expression is maintained by the transcriptional activators CLOCK (circadian locomotor output cycles kaput) and BMAL (brain and muscle aryl-hydrocarbon receptor nuclear translocator-like) and the transcriptional repressors PER (period) and CRY (cryptochrome). Also in the regulatory network are the retinoic acid-related orphan nuclear receptors REV-ERBα and RORα [118]. These molecules are present in the central nervous system as well as peripheral tissues. The impact of circadian rhythms on obesity and fatty liver disease is under active study [119]. Several master metabolic genes are regulated by the clock, including the PPAR gamma co-activators PGC1α and PGC1β and genes encoding fatty acid uptake and export [119].
Although the classical input that regulates the diurnal clock in all tissues is the availability of light, additional forms of input such as feeding can affect clocks in peripheral tissues. One intriguing observation arising from research on the circadian control of metabolism is that round-the-clock feeding of a high-fat diet induces obesity and NASH in mice, whereas the same diet given on a 12-h schedule does not, even if an identical number of calories is consumed [120]. More recently, it was reported that mice with pre-existing obesity can be cured of their dysmetabolism and hepatic steatosis by simply restricting their daily access to food to a 12-h cycle per day [121]. Although more research is necessary to understand the mechanism underlying this phenomenon, the observation is likely to prompt studies in humans with NAFLD and NASH.
Pathogenesis of Liver Injury in NASH
The term NASH, in contrast to NAFLD, connotes the presence of hepatic steatosis with accompanying liver cell injury and hepatic inflammation. Depending on the severity of disease, NASH can also feature hepatic fibrosis and even liver cancer. Hepatic steatosis is at the heart of both NAFLD and NASH; consequently, there is a tendency to assume these two entities form a continuum in which disease progresses from steatosis to steatohepatitis. Indeed, this is the basis for the “two-hit” hypothesis, in which the first hit to the liver is fat accumulation and a second hit, which can take many forms, triggers liver damage [122]. Although the two-hit concept has validity, there is also reason to consider that NASH represents the consequence of multiple parallel hits to the liver [123]. In this paradigm, NAFLD does not inevitably become NASH, and NASH may not be preceded by the more benign NAFLD. The discussion that follows does not specifically enlighten this debate but does address the cellular mechanisms of liver injury in NASH. The focus is on how liver cells are damaged by lipid accumulation and how resident liver cells and recruited inflammatory cells react to this damage to stimulate inflammation, fibrosis, and cancer.
Mechanisms of Cell Death in NASH
Death Receptor Induction and Activation
Death receptors, which are members of the TNF receptor superfamily, figure prominently in the pathogenesis of NASH (Fig. 4.6a). Death receptors implicated in NASH development include Fas/CD95 (Fas), the TRAIL receptor DR5, and the TNF receptor TNFR1. One of the earliest observations linking death receptors to fatty liver disease was the discovery that NASH patients express high levels of the Fas receptor on hepatocytes [124]. This was followed by the demonstration that fatty acids upregulate Fas expression by cultured hepatocytes and sensitize them to death from Fas agonists [125]. Fatty acids appear to regulate Fas through a unique mechanism involving c-Met, the receptor for hepatocyte growth factor (HGF). In normal hepatocytes, Fas and c-Met exist as a complex on the hepatocyte surface, which serves to sequester Fas from undesired activation (Fig. 4.6b) [126]. When hepatocytes are exposed to fatty acids, the Fas–c-Met complex dissociates. At the same time, fatty acids stimulate hepatocytes to produce Fas ligand (FasL); this permits an autocrine Fas–FasL interaction and promotes cell death [127]. In vivo, the importance of Fas to fatty liver disease has been documented using several independent approaches. For example, a 12-mer peptide of c-Met designed to bind and inhibit Fas protects against experimental fatty liver disease [127], whereas hepatocyte-specific deletion of c-Met, which leaves Fas uninhibited on the hepatocyte surface, enhances experimental steatohepatitis [128]. Recently, experiments have shown that the Fas receptor can also interact with the EGF receptor (EGFR) on hepatocytes. Contrary to c-Met, however, the EGFR is not complexed to Fas under basal conditions (Fig. 4.6b). Rather, the two proteins are normally separate, and fatty acids promote their interaction [129]. Insulin can signal through the EGFR to promote cell proliferation [130]. When EGFR binds Fas, however, insulin responses change from proliferative to cytotoxic. This is intriguing because it implicates insulin as a direct hepatotoxic molecule in a fatty liver.
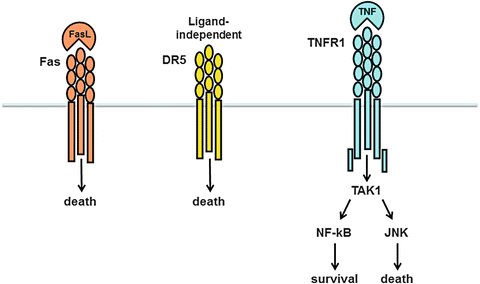
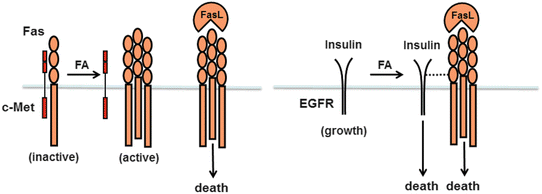
Fig. 4.6
Death receptors implicated in the pathogenesis of fatty liver disease. (a) Three different types of death receptors play a role in NAFLD and NASH: the Fas receptor, the TRAIL receptor DR5, and the TNF receptor TNFR1. Fas and its binding partner, Fas ligand (FasL), are both upregulated in fatty liver disease. DR5 expression is induced in hepatocytes in experimental fatty liver disease; receptor abundance leads to ligand-independent oligomerization and activation. TNFR1 can be activated on hepatocytes in a fatty liver but has the potential to signal either survival or death, via NF-κB or JNK, respectively. The outcome of TNFR1 activation depends on the balance between the two signaling processes and other simultaneous signaling processes within the cell. (b) The Fas receptor is subject to two unique types of regulation. First, Fas can exist in a complex with c-Met, which inhibits Fas signaling. Fatty acids stimulate dissociation of Fas from c-Met and permit cytotoxic signaling. Fatty acids can also stimulate a new association between Fas and the EGF receptor (EGFR). In this instance, EGFR signaling changes from growth to death. Thus, in the presence of fatty acids, both FasL and insulin (a ligand for EGFR) may signal cell death
TRAIL (TNF-related apoptosis-inducing ligand) is classically known as a tumoricidal agent [131]. TRAIL receptors, however, are also expressed on nonmalignant cells including hepatocytes, where they can stimulate cell death [132]. In the setting of fatty liver disease, the TRAIL receptor DR5 is upregulated on hepatocytes [133, 134]. Fatty acids upregulate the expression of DR5 in hepatocyte culture through a mechanism involving induction of p53 [134] or ER stress [135]. Importantly, enhanced expression of DR5 on hepatocytes can lead to cell death even in the absence of the TRAIL ligand (“ligand-independent activation”), so there is no need for an external stimulus. Hepatocytes lacking DR5 are resistant to fatty acid toxicity [135]. DR5-deficient mice also are protected against steatohepatitis in vivo, although it is currently unclear whether their improved outcome is directly attributable to reduced levels of hepatocyte death [136].
TNF is produced by dysfunctional adipose tissue [7, 8] and is present in the circulation of obese individuals [137, 138]. The cytokine is also produced locally within a fatty liver, as will be discussed below (see section “Mechanisms of Hepatic Inflammation”). Although hepatocytes express the death receptor TNFR1, TNF does not typically kill hepatocytes because the receptor signals multiple responses, not all of which lead to death (Fig. 4.6a). One of the top-level molecules activated by TNF is the MAP3 kinase TAK1 (transforming growth factor β-activated kinase 1). TAK1 in turn activates several downstream kinases, including Jun N-terminal kinase (JNK) and NF-κB. JNK activation typically promotes cell death (see section on lipotoxicity below), whereas NF-κB activation typically promotes survival. When normal hepatocytes are treated with TNF, the NF-κB-dependent survival signals typically dominate. In situations where NF-κB responses are compromised, however, the balance is upset, and TNF induces hepatocyte death [139]. This is relevant to NAFLD because studies in mice indicate that high-fat feeding suppresses hepatic expression of TAK1 [140]. This could certainly limit the survival response to TNF, but theoretically would also reduce TNF-mediated activation of JNK. Importantly, JNK is not completely dependent on TAK1 for activation in response to TNF (see below), and thus, the balance of TNF signaling in a fatty liver could be tipped toward death.
As just mentioned, hepatocytes can be sensitized to TNF-induced death by means other than suppression of the NF-κB survival response. TNF also signals the production of reactive oxygen species (ROS), which stimulate JNK activation [141]. Consequently, any alteration that limits the control of ROS in hepatocytes could tip the balance in favor of JNK and lead to TNF-induced death. Pertinent to NASH, experimental studies have shown that diets enriched in cholesterol can compromise hepatocyte control of ROS by promoting accumulation of free cholesterol in hepatocyte mitochondrial membranes [142]. The excess cholesterol disrupts membrane fluidity, which permits the loss of an important mitochondrial antioxidant, glutathione. Notably in humans with NAFLD and NASH, free cholesterol levels are increased in the liver [117], and genes regulating cholesterol synthesis are induced [143]. Together, these observations provide a rationale for the involvement of TNF as an important mediator of cell death in NASH.
Downstream Consequences of Death Receptor Activation: Apoptosis and Necroptosis
The engagement of death receptors on hepatocytes sets into motion a complex series of events that culminate in cell death. Death receptor activation can stimulate different modes of cell death depending upon the signaling pathway taken inside the cell. The classical response to death receptor engagement is apoptosis : this is a non-lytic form of cell death marked by an initial activation of caspase-8 and subsequent activation of “executioner” caspases including caspase-3, caspase-6, and caspase-7 [144] (Fig. 4.7). Death receptor activation can also induce a lytic form of death termed necroptosis [145]. In this instance, the initial intracellular event is activation of a complex between two receptor-interacting proteins (RIPs), RIP1 and RIP3. This leads to activation of mixed lineage kinase domain-like protein (MLKL), which perturbs the permeability of the plasma membrane leading to cell swelling and eventual rupture [146]. Notably, the RIP1/RIP3 complex is activated only when caspase-8 is inhibited, and thus, the two pathways are mutually exclusive within a single cell [145]. Both forms of death, however, appear relevant to NASH. In humans with NASH, activation of caspases is evident histologically and biochemically via the release of caspase-cleaved fragments of cytokeratin-18 into the circulation [147]. RIP3 is also upregulated in the livers of patients with NASH [148]. Studies in mice indicate that inhibition of caspases, either chemically or by genetic deletion, improves liver injury in experimental steatohepatitis [71, 149]. Likewise, RIP3 deletion ameliorates experimental liver injury in a mouse model of fatty liver disease [148]. Importantly, the blockade of either caspases or RIP3 alone does not completely reverse experimental steatohepatitis. This underscores that hepatocytes can follow more than one pathway to death in a fatty liver.
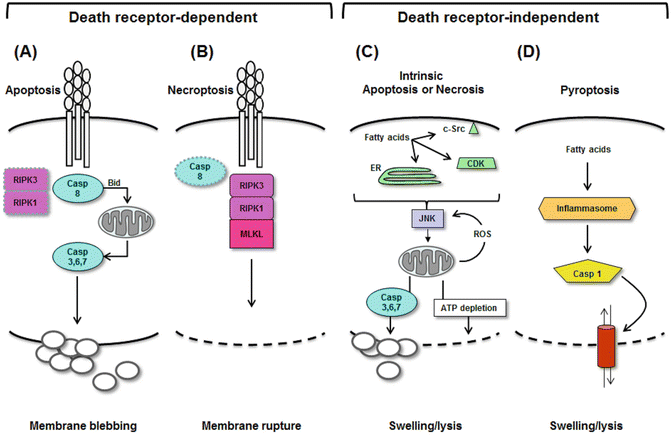
Fig. 4.7
Modes of hepatocyte death in NASH. Liver cells can take several routes to death in the setting of fatty liver disease. The modes of death can be segregated into death receptor-dependent and death receptor-independent processes. Activation of death receptors signaling either apoptosis or necroptosis. (a) Apoptosis occurs when death receptors activate caspase-8 (and inactivate RIP1/RIP3), leading to downstream activation of caspase-3 and condensation/blebbing of the plasma membrane. (b) Necroptosis occurs when death receptors are activated, but caspase-8 is inhibited, leading to downstream activation of RIP1/RIP3. This in turn leads to activation of MLKL, with subsequent lysis of the hepatocyte plasma membrane. (c) Fatty acids can kill cells independent of death receptors by causing intrinsic mitochondrial damage via JNK. Fatty acids can activate JNK by several mechanisms, including ER stress or activation of c-Src or cyclin-dependent kinases (CDK). JNK, once activated, binds to mitochondria and promotes ROS release. This begins a vicious cycle of JNK activation and oxidant stress that ultimately depletes cellular ATP, leading to necrosis. JNK can also cause apoptosis by stimulating mitochondria to activate caspase-3. In this case, JNK activation results in apoptosis. (d) Fatty acids can induce pyroptosis by upregulating inflammasomes and activating caspase-1. This opens a plasma membrane pore, leading to cell swelling and lysis
Death Receptor-Independent Apoptosis
Apoptosis triggered by death receptor activation is considered “extrinsic.” Another pathway to apoptosis, termed “intrinsic,” bypasses death receptors but still culminates in the activation of executioner caspases. Certain lipids, particularly the long-chain saturated fatty acids palmitate (C16:0) and stearate (C18:0) and their downstream metabolite lysophosphatidylcholine, are capable of inducing intrinsic apoptosis of hepatocytes through a process called lipotoxicity or lipoapoptosis [150–153]. In cell culture, an important prerequisite to hepatocyte lipotoxicity is fatty acid-induced activation of JNK [150]. In vivo as well, JNK activation is critical to the development of experimental NASH [154, 155] and has been documented in the livers of humans with NASH [117, 156]. One means by which saturated fatty acids can activate JNK is through ER stress [157, 158] (Fig. 4.7). During ER stress, JNK is activated downstream of IRE1 [159]. It then migrates to the mitochondrion where it perturbs mitochondrial respiration, prompting the release of mitochondrial cytochrome c and activation of caspase-3 [160]. JNK can also interfere with mitochondrial function indirectly, by activating pro-apoptotic proteins of the BH3-only family and inactivating their inhibitors in the Bcl-2 family, leading to a similar result [150, 161, 162]. Saturated fatty acids are also capable of activating JNK independently of ER stress. These alternative pathways involve cyclin-dependent kinases [163] or c-Src [164].
Since ER stress has been invoked as an important intermediate in fatty acid-induced cell death, it should be noted that ER stress can also lead to apoptosis through activation of PERK. PERK activation induces the expression of C-EBP homologous protein (CHOP), which in turn stimulates the transcription of pro-apoptotic proteins and suppresses the transcription of anti-apoptotic proteins [165, 166]. Although ER stress-induced upregulation of CHOP is an important intermediate to cell death in many cell systems [167, 168], it appears dispensable to fatty acid-induced death of hepatocytes [169, 170]. CHOP also induces the expression of ER oxidoreductin-1 (ERO1), an enzyme that promotes protein oxidation in the ER and contributes to cellular oxidant stress [171]. This sensitizes cells to death, although it does not appear to intersect directly with mitochondrial dysfunction and intrinsic apoptosis.
Pyroptosis as a Potential New Mode of Cell Death in Fatty Liver Disease
The term pyroptosis , which connotes a pro-inflammatory (“fiery”) form of programmed cell death, was coined in 2001 [172]. Pyroptosis , like apoptosis, involves caspase activation, but is dependent specifically on caspase-1 rather than caspase-3, caspase-6, caspase-7, or caspase-8. Pyroptosis is a lytic form of cell death; it is induced when caspase-1 is activated by a multi-protein complex called the inflammasome (discussed below) (Fig. 4.7). Although the principal function of caspase-1 is to promote inflammation, it can also lead to cell death by causing pore formation in plasma membranes [173]. In the livers of experimental animals, forced activation of caspase-1 via the inflammasome causes substantial hepatocyte injury [174]. With respect to the role of pyroptosis in fatty liver disease, studies show that saturated fatty acids stimulate hepatocytes to express inflammasome components in cell culture [175] and caspase-1 is activated in experimental NASH [175, 176]; importantly, upregulation of caspase-1 has also been documented in humans with NASH [175]. Whether pyroptosis is an important mode of hepatocyte death in fatty livers is still uncertain, because caspase-1 inactivation in experimental fatty liver disease has little effect on markers of hepatocyte injury [175, 177, 176]. Recent studies indicate caspase-1 does play a role in hepatic inflammation and fibrosis in experimental fatty liver disease (see section “Mechanisms of Hepatic Fibrosis”).
Mitochondrial Dysfunction and Necrosis
Necrosis is an unregulated form of cell death in which profound mitochondrial dysfunction leads to depletion of cellular ATP, failure of cellular ATP-dependent ion pumps, and plasma membrane rupture [178]. A decline in mitochondrial dysfunction is an important feature of NAFLD and NASH [179], and thus, some of the hepatocyte death that occurs in fatty livers is likely due to necrosis. Mitochondrial deterioration in fatty liver disease results from sustained high levels of fatty acid oxidation; this is driven in part by the continuous influx of fatty acids from adipose tissue and in part by the need for energy to drive processes such as lipogenesis and gluconeogenesis [39, 180]. The constant flux of fatty acids through mitochondria, with attendant high-level activity of the tricarboxylic acid (TCA) cycle [181], leads to enhanced delivery of electrons to the mitochondrial respiratory chain (MRC) and electron leakage within the MRC [182, 183]. This generates harmful ROS, which can damage the components of the MRC [184] and lead to more ROS production. ROS can further exacerbate mitochondrial dysfunction by activating JNK [160]; eventually this vicious cycle will result in mitochondrial failure and ATP depletion. It is important to note that in hepatocytes, mitochondrial dysfunction is central to all types of cell death [185], and thus, distinguishing among the specific types of cell death can be difficult (Fig. 4.7). However, because of the indolent nature of NASH compared to other liver diseases defined by necrosis, such as acetaminophen toxicity and ischemia–reperfusion injury, one can deduce that necrosis is not the predominant mode of cell death in a fatty liver.
Contribution of Impaired Autophagy to Cell Death in NASH
Autophagy is a process that typically promotes cell survival. Thus, any impairment of autophagy can make a cell vulnerable to death. In hepatocytes, autophagic breakdown of hepatic lipids is important for supplying hepatocyte mitochondria with fuel; consequently, any reduction in autophagy can lead to impaired mitochondrial ATP production and cell death. Impaired autophagy in a fatty liver can also lead to cell death by other means. For example, autophagy is the process used to rid a cell of dysfunctional mitochondria [186]. Since the metabolic demand placed on mitochondria in a fatty liver leads to dysfunction, any impairment in the ability of hepatocytes to remove damaged mitochondria may lead to cell death. Impaired autophagy can also sensitize hepatocytes to TNF-induced cell death by enhancing JNK signaling and stimulating caspase activation [187]. In addition, recent work indicates there is a reciprocal relationship between autophagy and ER stress in hepatocytes [188, 189]. In this case, impaired autophagy could enhance ER stress-induced lipotoxicity. Evidence of impaired autophagy has been found in the livers of NASH patients [188, 190]. This has led to interest in testing drugs that stimulate autophagy as a treatment for fatty liver disease. Experimental data are limited but promising [191, 189].
Acidic Sphingomyelinase and NASH
Acidic sphingomyelinase (ASMase) is an enzyme that converts sphingomyelin to ceramide and phosphorylcholine. A growing body of work points to a specific role for this enzyme in the pathogenesis of NASH [192]. The ceramide generated by ASMase can facilitate TNF-induced hepatocyte apoptosis [193]; it can also inhibit the synthesis of phosphatidylcholine, which as mentioned earlier is important to hepatic lipid export as well as the integrity and function of organelle membranes [194]. Importantly, ASMase inhibits methionine adenosyltransferase [195]. This enzyme is responsible for converting methionine to S-adenosylmethionine, a critical methyl donor and glutathione precursor [196]. Thus, ASMase is integrally linked to processes pertinent to NASH, ranging from ER stress to autophagy to cell death and oxidant stress. Studies in mice indicate that genetic deletion of ASMase protects against experimental NASH [189]. Pharmacologic inhibition of ASMase with the drug amitriptyline is also effective [189], and thus, the role of this enzyme in human NASH warrants further exploration.
Mechanisms of Hepatic Inflammation in NASH
Activation of the innate immune system is a key feature of NASH. A number of different compounds can elicit innate immune responses in a fatty liver; some originate outside the liver and reach the organ from the circulation, whereas others are produced locally and act in paracrine or autocrine fashion. One external stimulus to hepatic inflammation in NASH is gut-derived bacterial endotoxin. The role of endotoxin in the pathogenesis of NASH has been established through a variety of experiments demonstrating that prebiotics, probiotics, and antibiotics, by reducing endotoxin delivery to the liver, can interfere with NASH development in experimental animals [197–199]. Second, fatty acids, which are abundant in the liver as well as the circulation in obese individuals with hepatic steatosis, can induce innate immune responses [200, 201]. Third, molecules released locally inside the liver by dead or dying hepatocytes serve as a stimulus to innate immune activation. Collectively these diverse compounds induce inflammation by acting either as PAMPs (pathogen-associated molecular patterns) or DAMPs (damage-associated molecular patterns), which engage pattern recognition receptors (PRRs) on resident liver cells.
Toll-Like Receptors in the Pathogenesis of Hepatic Inflammation
The PRRs that prompt hepatic inflammation in NASH belong to the Toll-like receptor (TLR) family. Engagement of TLRs triggers inflammation by initiating a signaling cascade that activates NF-κB and culminates in the transcription of genes encoding pro-inflammatory cytokines. TLRs are expressed by all resident liver cells [202]; consequently, the liver has broad potential to respond to PAMPs and DAMPs through this mechanism. Among all the hepatic TLRs, TLR2, TLR4, and TLR9 are the ones implicated in the pathogenesis of steatohepatitis (Fig. 4.8). Traditionally, TLR2 classically recognizes peptidoglycans and other components of Gram-positive bacteria, whereas TLR4 recognizes Gram-negative endotoxin and TLR9 binds DNA sequences found in bacteria and DNA viruses [203]. Accordingly, it is logical that TLR4 as the endotoxin receptor would play a role in NASH-related inflammation, and mice lacking TLR4 are in fact resistant to diet-induced NASH [204, 205]. Roles for TLR2 and TLR9 are less intuitive, based on the seeming lack of appropriate ligands for these receptors. Recent data, however, indicate that TLR9 recognizes not only bacterial DNA but also mammalian DNA [206]. This makes TLR9 a potential receptor for DNA released from dying liver cells, and mice deficient in TLR9 are also protected from experimental NASH [207]. Making even better sense of TLRs in NASH is emerging evidence that TLRs 2, 4, and 9 are all capable of recognizing compounds released by injured liver cells [208]. That said, the influence of TLR2 on NASH is currently controversial. Although a role for TLR2 has been suggested based on improvement of experimental NASH in TLR2 knockout mice [209], others have observed that mice lacking TLR2 actually develop worse NASH [204]. The exact contribution of this TLR to inflammation in fatty livers is yet to be determined.
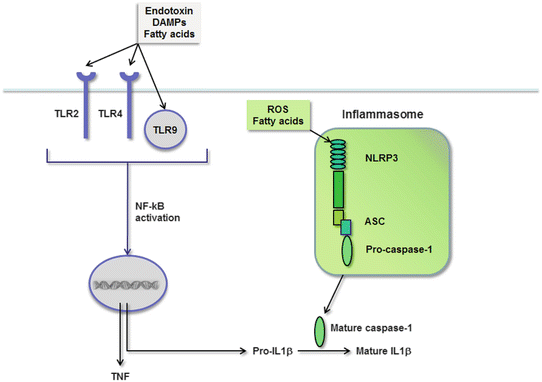
Fig. 4.8
TLRs and the inflammasome signal hepatic inflammation in NASH. TLRs are activated by fatty acids, endotoxin, and DAMPs, which leads to NF-κB activation and the production of pro-inflammatory cytokines such as TNF and IL-1β. TNF is active upon synthesis, but IL-1β is produced as an inactive propeptide. Fatty acids and ROS also stimulate the synthesis and assembly of the NLRP inflammasome, comprised of NLRP3, ASC, and caspase-1. This complex facilitates the cleavage and activation of IL-1β
Just as endotoxin and DAMPs can bind TLRs in a fatty liver, so can fatty acids. This is particularly true of long-chain saturated fatty acids, which bear structural resemblance to the lipid A moiety of bacterial endotoxin, making them ligands for TLR4 [210]. Saturated fatty acids can contribute to hepatic inflammation in NASH by other means as well, as discussed below.
Role of the Inflammasome in Steatohepatitis
As mentioned previously, TLR activation triggers inflammation by stimulating the production of pro-inflammatory cytokines. Prominent among the cytokines induced in response to TLR engagement are TNF and IL-1β. Once synthesized, TNF can be readily secreted from the cell; IL-1β, on the other hand, cannot be secreted until it is first processed from a propeptide to a mature form. The enzyme responsible for cleaving IL-1β is caspase-1. Caspase-1 itself is initially inactive inside the cell and must be activated by proteolytic cleavage within a multimolecular complex called the inflammasome [211] (Fig. 4.8).
Inflammasomes are critical participants in inflammatory responses in a wide variety of cells and tissues. In the majority of cases, they consist of nucleotide-binding oligomerization domain (NOD)-like receptor proteins (NLRPs) that sense an activating signal, adaptor proteins termed ASC (apoptosis-associated speck-like protein containing a caspase activation and recruitment domain), and procaspase-1 [211]. NLRPs come in many forms, but the one capable of responding to the broadest range of activating signals is NLRP3. The cells typically equipped with inflammasome components are innate immune cells, including macrophages, neutrophils, and dendritic cells [211–214]. In the liver, Kupffer cells as resident macrophages possess all the machinery of the NLRP3 inflammasome; NLRP3 components have also been identified, however, in hepatocytes, sinusoidal endothelial cells, and hepatic stellate cells, which creates the potential for an expanded role for inflammasome complexes in liver disease. Evidence indicates that inflammasomes are associated with steatohepatitis, based on the induction of inflammasome components or the presence of active caspase-1 or active IL-1β in the livers of animals and humans with NASH [215, 207, 175, 216, 177, 176, 217, 218]. More importantly, inflammasome activation appears to directly influence the development of steatohepatitis, as targeted disruption of inflammasome components in animal models of NASH reduces IL-1 secretion and limits the severity of diet-induced hepatic inflammation [218, 177, 176].
Until recently, inflammasome-mediated cytokine activation was believed to require two independent signals: one to stimulate cytokine production and a second to stimulate assembly of the inflammasome complex [208]. Cytokine production would occur downstream of a TLR signal, whereas inflammasome activation would result from one of a number of cellular stresses. Emerging data, however, indicate that certain DAMPs can serve both functions [219, 220]. In the context of fatty liver, saturated fatty acids are in a position to singlehandedly activate IL-1β via the inflammasome. Not only can they bind TLR4 to stimulate cytokine production, but they can also activate NLRP3 by inducing intracellular oxidant stress [200, 201]. The prospect that fatty acids alone might completely activate the inflammasome is intriguing, but has not been proven experimentally. Indeed, studies to date in liver cells indicate that fatty acids do not activate the inflammasome without a second signal [175, 209]. Another provocative concept is that ER stress may act as a dual stimulus capable of stimulating IL-1β synthesis and activating the NLRP3 inflammasome [221].
Contribution of Kupffer Cells to Hepatic Inflammation in NASH
Although several types of leukocytes have the potential to contribute to innate immune activation in the liver, Kupffer cells are key mediators of hepatic inflammation in NASH. These cells are uniquely positioned in the hepatic sinusoids to be the first responders to PAMPs and DAMPs, and they are robust producers of IL-1 and TNF. Kupffer cell-derived IL-1 and TNF can exacerbate hepatic steatosis by stimulating fat accumulation within hepatocytes [207, 215]; IL-1 and TNF can also kill steatotic hepatocytes and prompt the release of more DAMPs [142, 207, 222]. In addition, Kupffer cells produce chemokines such as CCL2 and RANTES (regulated on activation, normal T cell expressed and secreted) [223] that recruit inflammatory monocytes from the circulation into the liver. Collectively, therefore, Kupffer cells contribute to both the development and perpetuation of injury and inflammation in a fatty liver. Studies in mice indicate that Kupffer cell activation is critical to the initiation of hepatic inflammation in NASH. Specifically, if Kupffer cells are eliminated from the liver in the early phase of experimental fatty liver disease, cytokine production is reduced, and the recruitment of inflammatory cells to the liver is significantly attenuated [224]. Once the inflammatory process is set into motion, however, the role of Kupffer cells in NASH becomes less critical, presumably because recruited monocytes perform many of the same functions.
The mononuclear cells that invade an injured liver tend to have a more inflammatory phenotype than resident Kupffer cells [225]. Infiltrating mononuclear cells are often described as having an M1, or pro-inflammatory, phenotype as opposed to an M2 phenotype, which is anti-inflammatory [226]. M2 Kupffer cells have the ability to kill M1 cells and also to promote hepatocyte survival [227, 228]. This suggests that M2 cells are equipped to maintain homeostasis in the normal liver. M2 Kupffer cells can adopt an M1 phenotype upon activation, and the degree to which this occurs may be an important determinant of the severity of NASH. In experimental models of steatohepatitis, the severity of liver injury is often strain dependent, and this variation has been attributed to differences in Kupffer cell M1/M2 balance [229, 227]. This has generated interest in identifying factors that enhance M2 polarization; putative agents include unsaturated fats, which can activate peroxisome proliferator-activated receptor-δ (PPARδ), adiponectin, or ligands for the cannabinoid receptor CB2 [230–232].
Mechanisms of Hepatic Fibrosis in NASH
Liver fibrosis develops in NASH following a sustained period of hepatocyte injury and organ inflammation. This scenario is typical of many chronic liver diseases, and thus, it is not surprising that some of the mechanisms promoting liver fibrosis in NASH are shared with other disease processes. The hallmark of hepatic fibrosis is the priming and activation of hepatic stellate cells to a myofibroblastic phenotype. Several events can lead to stellate cell activation in fatty liver disease.
Hepatocyte Death and Liver Fibrosis
One potent stimulus to stellate cell activation is the death of hepatocytes. Specifically, fragments of dead hepatocytes can be ingested by stellate cells, which triggers the transformation of stellate cells to myofibroblasts [233]. Hepatocyte death is considered an important fibrotic stimulus in fatty liver disease because pan-caspase inhibitors, which inhibit hepatocyte apoptosis, can suppress fibrosis in mice with experimental steatohepatitis [234]. Cell death may also trigger fibrosis indirectly in a fatty liver, by first stimulating hepatic inflammation through the innate immune pathways described above [71, 218]. In support of this concept is the fact that TNF and IL-1β, which are both induced in NASH-related hepatic inflammation, can promote liver fibrosis. Notably, these cytokines do not induce fibrosis by stimulating stellate cell activation, but instead facilitate stellate cell survival through the activation of NF-κB [235]. In addition, stellate cells express TLRs, which enables them to respond directly to PAMPs and DAMPs in the setting of NAFLD. In this respect, endotoxin and synthetic TLR2 ligands have both been identified as stellate cell activators. Saturated fatty acids, however, do not directly activate stellate cells [236, 209]. Overall, many of the same stimuli that promote inflammation in NASH also promote fibrosis, which may explain why fibrosis tends to improve in parallel with inflammation upon therapeutic manipulation [207, 209, 237–239].
Hepatocyte “Un-death” and Liver Fibrosis
One of the hallmarks of NASH in humans is the presence of ballooned hepatocytes [240]. These cells are believed to have started down a death pathway, but stalled in the process, hence the designation “undead” [241]. Research into the phenomenon of ballooning has revealed that ballooned cells produce sonic hedgehog [242, 243, 241], a protein that promotes tissue fibrosis [244]. Hedgehog appears to mediate fibrosis indirectly by first inducing stimulating cholangiocytes to produce osteopontin. Osteopontin then acts in a paracrine fashion on stellate cells to induce fibrosis [245]. Experiments indicate that hedgehog inhibitors improve fibrosis in experimental fatty liver disease [246]), which supports the concept that hedgehog is an important contributor to NASH-related liver fibrosis. There remains some debate, however, whether hedgehog signaling specifically stimulates hepatic fibrosis or plays a role in upstream disease processes as well, such as hepatocyte death [247].
Influence of Adipokines on Liver Fibrosis
Some compounds with relevance to obesity and the metabolic syndrome have unique influences on stellate cells. Leptin , for example, which is abundant in obesity, interacts directly with HSC to stimulate collagen production [248]. Adiponectin , by contrast, which is scarce in individuals with the metabolic syndrome [249], suppresses stellate cell activation [250]. Thus, the adipokine profile of patients with NAFLD/NASH is one that favors hepatic fibrogenesis. Other adipokines, including resistin, visfatin, and apelin, correlate positively with NASH and liver fibrosis, but their roles in stellate cell activation are less well defined (reviewed in [251]).
Hepatocarcinogenesis in NASH
Just as chronic liver injury predisposes to hepatic inflammation and fibrosis, it also facilitates the development of liver cancer. A recent review [252] provides an excellent overview of the signaling pathways relevant to hepatocarcinogenesis in NASH. Observations in experimental animals have highlighted a role for NAFLD as an accelerant to liver cancer; specifically, they have shown that a carcinogenic insult is much more potent when administered on a background of obesity and fatty liver [253–255]. In one instance, mice fed a high-fat diet and then treated with the carcinogen diethylnitrosamine (DEN) developed liver tumors of twice the number and twice the size as mice given DEN with a standard diet [255]. Two factors implicated in the acceleration of tumor growth in the obese mice were IL-6 and TNF. These cytokines, which are both known to be upregulated in obese mice, enhanced hepatic activation of the stress kinases JNK and ERK and accentuated hepatic steatosis and inflammation in response to DEN.
The discovery that obesity and fatty liver enhanced JNK activation in the livers of obese DEN-treated mice is of interest because liver cancer arises in other animals that exhibit an imbalance between JNK (death) signaling and NF-κB (survival) signaling in hepatocytes in response to TNF [256] (Fig. 4.6a). More recently, researchers discovered another important driver of hepatocarcinogenesis downstream of TNF, mTOR. As mentioned earlier (see section “Death Receptor Induction”) JNK and NF-κB are activated by TNF through the MAP3 kinase TAK1. Importantly, TAK1 also activates AMP kinase, which then phosphorylates and inactivates mTOR [257]. In genetic and diet-induced obesity, however, TAK1 is suppressed [140]; this leads to abnormal mTOR activation, with an attendant increase in hepatic lipogenesis, decrease in hepatocyte autophagy, and decrease in fatty acid oxidation. In the extreme case of genetic ablation of TAK1, high-fat feeding induces steatohepatitis, hepatic fibrosis, and tumor formation, all of which are inhibitable by the mTOR inhibitor rapamycin. This implicates mTOR as an important driver of hepatocarcinogenesis in conjunction with JNK/NF-κB imbalance [140].
Yet another intriguing finding from a recent animal study is that ER stress in hepatocytes predisposes to liver cancer. What made this study unique was that ER stress was induced only transiently in mice, before the initiation of a high-fat diet. The mice that underwent, but then recovered from, an initial ER stress subsequently developed accentuated ER stress in response to high-fat feeding. This resulted in greater hepatic steatosis, more cell death, and more liver tumors [258]. This raises the possibility that a previous hepatic insult sensitizes a liver to metabolic damage, including liver cancer. In this respect, prior ER stress can serve as a risk factor for enhanced liver damage, rather than a preconditioning event that would suppress subsequent injury.
Summary
As knowledge about cell biology advances, so does our concept of the pathogenesis of fatty liver disease. This is immediately evident in the fact that we now include ER stress, autophagy, and inflammasome activation among the pathways contributing to hepatic steatosis and NASH. These new paradigms are not replacing old theories; rather, they are putting them into new perspective and in many cases permitting consolidation of seemingly unrelated events into single pathogenic schemes (e.g., consider fatty acids, dead cells, and endotoxin all as inducers of the inflammasome in a fatty liver). Although for simplicity, specific disease mechanisms were often addressed individually in this chapter, they frequently intersect. For example, there is crosstalk between ER stress and autophagy [259], and JNK activation can result from numerous insults including ER stress, death receptor signaling, and ROS induction. This underscores the true complexity of disease pathogenesis in a fatty liver.
With regard to the pathophysiology of NASH and its complications, one theme that emerges from the above discussion is the central role of hepatocyte death in the process. Indeed, hepatocyte death triggers not only inflammation but also fibrosis and even cancer. With this in mind, it remains a conundrum why some individuals at risk for fatty liver disease develop only hepatic steatosis, whereas others develop NASH. Further exploration of genetic and behavioral modifiers is likely to shed light on this issue.
Grant Support
R01 DK068450, P30 DK026743.
Conflict of Interest
The author has no conflicts to disclose.
References
1.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante Jr AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808.PubMedCentralPubMed
2.
Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest. 2003;112(12):1785–8.PubMedCentralPubMed
3.
Trayhurn P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol Rev. 2013;93(1):1–21. doi:10.1152/physrev.00017.2012.PubMed
4.
Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46(11):2347–55.PubMed
5.
Alkhouri N, Gornicka A, Berk MP, Thapaliya S, Dixon LJ, Kashyap S, et al. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J Biol Chem. 2010;285(5):3428–38. doi:10.1074/jbc.M109.074252.PubMedCentralPubMed
6.
Gornicka A, Fettig J, Eguchi A, Berk MP, Thapaliya S, Dixon LJ, et al. Adipocyte hypertrophy is associated with lysosomal permeability both in vivo and in vitro: role in adipose tissue inflammation. Am J Physiol Endocrinol Metab. 2012;303(5):E597–606. doi:10.1152/ajpendo.00022.2012.PubMedCentralPubMed
Related posts:
 Nonalcoholic Fatty Liver Disease in Children
Diagnostic Considerations and Clinical End Points for Nonalcoholic Steatohepatitis
Hepatic and Extrahepatic Malignancies in NAFLD
Nonalcoholic Fatty Liver Disease in Children
Diagnostic Considerations and Clinical End Points for Nonalcoholic Steatohepatitis
Hepatic and Extrahepatic Malignancies in NAFLD
 Clinical Features, Disease Modifiers, and Natural History of Alcoholic Liver Disease
Clinical Features, Disease Modifiers, and Natural History of Alcoholic Liver Disease
 Pathogenesis of Alcoholic Liver Disease
Pathogenesis of Alcoholic Liver Disease
 Animal Models of Alcoholic Liver Disease
Animal Models of Alcoholic Liver Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


