1.Heavy and steady ethanol intake
2.Physical and metabolic tolerance to ethanol: achieving high BACs which are tolerated by the model
3.Chronicity of ethanol intake: at least 10~25 % of life span
4.Incorporation of genetic and environmental risk factors
5.Reproduction of clinically relevant, advanced ALD liver pathology
6.Reproduction of clinical symptoms/complications of ALD patients (portal hypertension, coagulopathy, hypoalbuminemia, hyperbilirubinemia, muscle wasting)
ALD is the disease which develops after 15–20 years of heavy drinking in man and 10–15 years in woman. This chronicity is an important consideration and means that in rodents, for example, 2–6 months of drinking in the quantities that maintain sustained BACs, must be required. The pattern of ethanol intake is also critical. As already discussed above, alcoholics continually take ethanol throughout the day to maintain BACs every 3–4 h between drinking episodes from morning through evening followed by a large dose before they sleep. Binge drinking pattern is commonly observed among youth [3], and binge superimposed to steady heavy drinking is frequently associated with AH [4, 5]. The model needs be able to test this chronic plus binge pattern. The model should also allow incorporation of genetic and environmental risk factors known for ALD patients [6]. This is particularly essential as research in the past 4 decades has led to a consensus that heavy ethanol intake is required but not sufficient alone to cause advanced ALD. Lastly, the model should reproduce not only advanced ALD spectra but also symptoms and complications associated with them, such as endotoxemia, bacteremia, jaundice, portal hypertension, and ascites, which are clinically and therapeutically important in dealing with ALD patients. Only then, the model will be realistically useful for understanding the mechanisms underlying complications and testing new therapeutic modalities. This review serves to critically analyze the animal models of ALD from the viewpoint of these requirements.
Historical Perspective
History repeats itself. Much can be learned from it to move forward and avoid unnecessary repetitions. This principle also applies to science and certainly for research on animal models of ALD. By reviewing the history, we realize that there are key messages to learn from the contributions made in the past. Historical descriptions summarized below are in part based on excellent reviews previously published [7–9] and will touch upon major developments and directions the field has experienced since 1930 (Fig. 5.1).
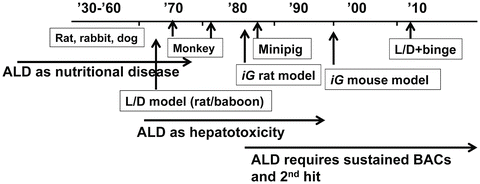
Fig. 5.1
Historical perspective. A schematic diagram depicting the times when different models were developed or used and different proposals were made for ALD pathogenesis. For simplicity, only selected models and hypotheses are highlighted
Earlier Attempts of Ethanol Gavage for Forced Administration
As early as the 1930s, investigators realized that animal’s natural aversion to ethanol is a major limiting factor, necessitating the use of stomach tube for ethanol administration—gavage ethanol administration. Connor and Chaikoff fed dogs a high-saturated-fat, high-protein diet (lard and lean meat) along with vitamin and salt mixtures for 30–35 days to generate fatty liver and then gave lean meat ad lib and ethanol via gavage (approx. 2 ~ 3 g/kg) twice daily for 4–7 days. This regimen was alternated with a similar period of feeding the lard/meat diet for up to 106 days. All 16 dogs that underwent this treatment developed severe fatty liver with hyaline degeneration, and 4 of 16 dogs showed cirrhosis resembling early fatty cirrhosis in man. Although an appropriate control group was missing, this study most likely was the first 2-hit experiment with high fat and ethanol [10]. Connor then took a similar approach of feeding a diet high in protein and fat supplemented with vitamins to rabbits and administered ethanol via gavage for 3–8 months. Some had developed cirrhosis which appeared to have resulted from portal-portal bridging fibrosis [11]. A common pitfall of these earlier studies was poor nutritional status as ethanol intoxication achieved by gavage prevented the animals from having normal feeding. Chey et al. performed a more controlled study by feeding dogs a beef diet and ethanol via surgically implanted gastric fistula 5 days a week for 10–18 months achieving sustained nutrition and BACs (315–477 mg%). Liver histology showed fatty liver, ballooned hepatocyte degeneration, zonal and spotty necrosis, inflammation, and centrilobular fibrosis, the changes comparable to those seen in pre-cirrhotic ALD in patients [12].
Rodent ALD Model: A Debate Over Nutritional vs. Hepatotoxic Disease
In the comprehensive review by Moon in 1934, he pointed out that the literature lacks evidence to support that ethanol is a direct cause of liver damage [13]. This was followed by the findings that rats given 20 % of ethanol in drinking water developed cirrhosis when they were fed a low-protein, low-choline diet but not when these dietary deficiencies were corrected by supplementations [14–16]. These results supported the notion first proposed by Kennedy in 1933 that dietary deficiency was the crucial etiological factor in ALD [17]. Ashworth conducted the first rat model study utilizing forced ethanol intake by stomach tube and ad lib diet feeding and concluded that fatty liver produced in the model was a direct toxic effect of ethanol [18]. But as in the rabbit studies described above, poor feeding and nutrition caused by ethanol gavage was problematic. In 1949, Best et al. introduced for the first time isocaloric pair feeding by substituting ethanol consumed by ethanol diet-fed mice with equal calories of sucrose. Ethanol-fed mice developed fatty liver, but it was prevented by supplementation with a methyl donor such as methionine, choline, or casein [19]. This finding supported the concept that ethanol-induced fatty liver is caused not by ethanol’s toxic action but by increased requirements for choline and other nutrients by ethanol. Subsequent studies substantiated this concept. In particular, a series of publications by Porta and Hartroft established the importance of lipotrope deficiency in exacerbation of ALD and induction of cirrhosis [20, 21]. Methionine- and choline-deficient diet commonly used for NASH studies now originated from these earlier studies that established the importance of the deficiency of these nutrients in promoting alcoholic cirrhosis. In 1963, Lieber et al. developed an ethanol-containing liquid diet which contained all required nutrients in adequate concentrations and a control diet in which ethanol was isocalorically substituted by carbohydrate [22]. This diet achieved ethanol intake of 12–18 g/kg/day, 36 % of caloric intake from ethanol, and postprandial BACs of 100–150 mg%, in young growing rats (1 month old weighing ~150 g). Fatty liver still developed in these rats, supporting the view that ethanol exerts toxic effects to cause liver pathology despite adequate nutrition [22, 23]. This hepatotoxicity hypothesis was mechanistically supported by the MEOS (microsomal ethanol-oxidizing system) that the Lieber laboratory described [24, 25] and was later identified to be CYP2E1 and CYP reductase [26, 27]. However, the Lieber-DeCarli (L/D) diet method was criticized for not providing adequate nutrition and optimal growth in growing rats [28]. This nutritional inadequacy resulted from limited intake of ethanol-containing diet due to the animal’s aversion to ethanol, equally limiting the intake of control diet by pair-fed control rats. Thus, even with adequate concentrations of macro- and micronutrients in the diet, intake of suboptimal quantities of the diet resulted in marginal nutritional status. In support of this notion, if ethanol content in the L/D diet was reduced to 26 % of calories from 36 % calories (the diet with more diluted ethanol), the volume of the ethanol diet consumed by rats increased to 50 %, still achieving comparable daily ethanol dose per mouse compared to the 36 % diet; the rats grew optimally [29], and alcoholic fatty liver was prevented [30].
Subhuman Primate Models
The nutrition vs. toxicity debate also involved primate models. The L/D diet was applied to baboons, and as they consumed as much as 50 % of calories from ethanol over 1–4 years, they developed fatty liver, steatohepatitis, perivenular and bridging fibrosis, and cirrhosis [31, 32]. A primary conclusion from these studies was that baboons tolerated higher ethanol intake and developed progressive ALD while rats given L/D diet had limited liver pathology due to lower ethanol intake and BACs. However, Macaca radiata monkeys [33], rhesus monkeys [34], and baboons [35] fed with L/D or similar diet did not develop advanced ALD as observed in Lieber’s studies.
Miniature Pig Model
Hanford miniature pigs have been fed low- or moderate-fat (5 % Cal or 12 % Cal) regular chow mixed with ethanol (4 g/kg/day, 40 % Cal) for 12 or 20 months to study alcoholic liver injury. Under these regimens, the average BAC levels of 130–229 mg% were measured; serum transaminase levels were modestly elevated, but liver histology was not altered [36]. However, when the corn oil content in the diet was increased to 33 % Cal in Yucatan miniature pigs, ethanol feeding resulted in fatty liver and liver fibrosis [37]. Two major advantages of this model are that (1) miniature pigs have anatomical, physiological, and immunological similarities with human and (2) miniature pigs have the voracious appetite with less aversion to ethanol or dietary manipulation as compared to other small animal models. Thus the model has the potential for further experimental manipulations including incorporation of the second or multiple hits that may lead to reproduction of advanced ALD. Obvious downsides of the model are longer periods of feeding and higher animal and per diem costs.
Sustained BACs by Intragastric Feeding (iG) Model
One hypothesis which emerged from the studies on the rat and primate models described above was the importance of sustained BACs in the ALD pathogenesis. This was a difficult requirement to fulfill in animal models. Animals with aversion to ethanol do not become sufficiently addicted to self-administered large quantities of ethanol to the degree seen in alcoholics, and animals have higher basal metabolic rates which directly translate to faster ethanol metabolism, making sustained and high BACs in animals especially in rodents almost impossible. This challenge served as an impetus for the development of intragastric ethanol feeding model in rodents. The iG model allowed excessive ethanol intake, sustained BACs (250–350 mg%), and controlled nutritional intake [38]. In adult rats, iG model achieved ~49 % Cal intake from ethanol and induced severe fatty liver even with a low-fat diet [38]. More advanced pathology such as liver necrosis and fibrosis was produced with high-fat diet [39] and cirrhosis with a combination of high-fat diet and iron [40], thus supporting the importance of both sustained BACs and the secondary risk factors in progressive ALD. In the 2000s, Thurman’s laboratory has performed a series of elegant mouse iG experiments to demonstrate the importance of CD14, ICAM-1, TLR4, and LBP in experimental ALD by performing genetic loss of function experiments [41–44]. This mouse iG model has evolved in the past 2 decades (Fig. 5.2) [45] and revealed the importance of clinically relevant 2nd hit such as hepatitis C virus protein expression [46] and obesity [47] which will be discussed below.

Fig. 5.2
Mouse iG model. (a) The iG catheter used for the model. (b) An iG mouse is placed within an individual micro-isolator cage. The catheter is connected to a flow-through swivel clamped above the cage. (c) A close-up view of the iG mouse. (d) An overview of the iG model setup
Three noteworthy observations were made in the iG models concerning their BACs and metabolic and physical tolerance. Firstly, the animals exhibited unexpected cyclical daily BACs with a magnitude of 50–450 mg% peaking every 5–6 days despite continuous iG infusion of a constant ethanol dose (Fig. 5.3a) [48]. This cycle was triggered by induction of ethanol clearance with the threshold BAC of ~250 mg%. This phenomenon was confirmed independently by other laboratories [49, 50] and mimicked the cyclical pattern of the amount of ethanol consumed by monkeys with iG self-administration (Fig. 5.3b) which simulated the pattern of ethanol consumption by alcoholics [51]. In the iG model which bypasses the cephalic and oral phases of intake, the rate of ethanol consumption is most directly governed by the rate of ethanol metabolism. Thus the exhibition of the cyclic BAC pattern must be metabolic in origin. If ethanol metabolism peaks every 5–6 days in man, this may underlie binge intake with a similar interval seen in alcoholics. The second observation was remarkable metabolic tolerance that the iG rodents underwent. The progressively increasing dose of ethanol was tolerated by these animals, depicting their remarkable metabolic reserve which was only unmasked by the iG method. This metabolic tolerance is matched by physical tolerance. Under the iG regimen, the BAC of 250 mg% or less does not cause any noticeable physical sign of intoxication. At the cyclical peak of BACs of 450–550 mg%, the animals exhibit suppressed motor coordination, particularly in lower part of the body, and only when BACs exceed 600 mg%, anesthetic effects of intoxication ensue. This remarkable physical tolerance develops because of heavy ethanol intake achieved in the iG model. Therefore, it seems that ethanol intake and intoxication facilitated by ad lib feeding methods reflect only mild level of drinking which does not reproduce heavy drinking of ALD patients.
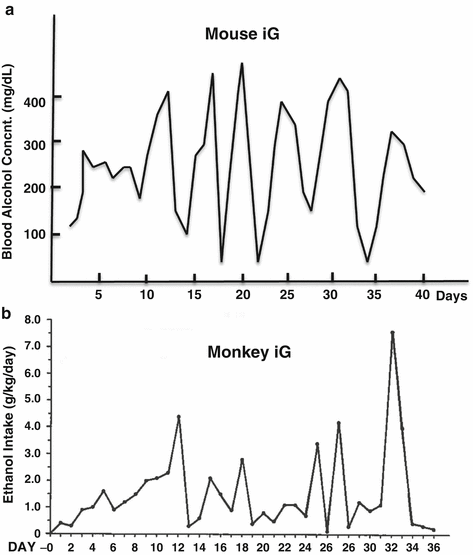
Fig. 5.3
Cyclical phenomenon . (a) Cyclic blood ethanol concentrations of an iG mouse despite the constant ethanol infusion. (b) A similar cyclic pattern is observed in the dose of ethanol consumed by a monkey who self-administered it via iG catheter (A figure with permission from Altshuler, H.L., Intragastric self-administration of ethanol: a subhuman primate model of ethanolism. In: Animal Models in Ethanol Research. Ed., Eriksson K, Sinclair JD, Kiianmaa K. Academic Press, New York, 1980, pp179–183.)
Chronic Ethanol Plus Binge
Stomach gavage has been used since the 1930s as the method of force-feeding ethanol to animals that otherwise consume less due to the aversion. When binge is given subsequently to 10 days of L/D diet feeding, neutrophilic infiltration is induced with elevated serum ALT [52, 53]. This rather simple model, termed “NIAAA model” [52], has been used to disclose the importance of E-selectin and invariant natural killer T cells in neutrophilic inflammation [53, 54]. Binge can also be repeated as seen in alcoholics, and this approach combined to the iG mouse model fed with Western solid diet ad lib resulted in alcoholic neutrophilic hepatitis in mice with clinical features of AH in patients [55] as discussed in detail below.
Comparisons of Different Rodent ALD Models
Rodents are convenient and common species used for ALD research. For this reason, innovations that make rodent ALD models more clinically relevant, useful, and versatile are important in the field. We have witnessed such developments in recent years and this section will highlight them in addition to providing a generalized comparative summary on the procedures, liver pathology outcome achieved and not achieved, and advantages vs. weaknesses of different rodent models used in the past and currently (Table 5.2).
Table 5.2
Rodent models available for ALD research
EtOH administration | Models | Duration | Species | ALT & AST (U/L) | Liver pathology | Comments and references | |||
|---|---|---|---|---|---|---|---|---|---|
Steatosis | Necrosis | Inflammation | Fibrosis | ||||||
EtOH in drinking water, ad lib | 20–40 % (v/v) | 7–29 w | Mouse Rat | ALT: ~68 AST: ~107 | ++ | + | ++ | −/+ | •Convenient but long duration |
Oral gavage (EtOH in water, 32–50 %, v/v) | Acute single binge (4–7 g/kg) | 4–72 h | Mouse | ALT: 4 h: ~116 12 h: ~74 24 h: ~31 72 h: ~32 | ND | ND | ND | ND | |
Two-hit model: acute single binge + LPS (5 mg/kg, i.v.) | 24 h | Rat | AST: ~779 ALT: ~760 | − | ++ | ++ | − | •Neutrophil infiltration •EtOH causes early tolerance and late sensitization of KC to LPS [61] | |
Subacute repeated binge (4–5 g/kg, every 12–24 h) | 3–4 d | Mouse Rat | ALT: ~45 | −/+ | − | − | − | •MtDNA depletion and impaired hepatic respiration [64] | |
Two-hit model: subacute repeated binge in obese rat | 3 d | Rat | ALT: ~125 | + | − | + | − | •Obesity aggravates liver injury through oxidative and nitrosative damage [65] | |
Chronic repeated binge (5 g/kg daily) | 8 w | Rat | ALT: ~120 AST: ~160 | + | + | + | − | •Elevation of LPS and KC activation [66] | |
Intravenous | Single i.v. injection (1.75 g/kg) followed by i.v. infusion (250–300 mg/kg/h) | 18 h | Rat | ALT: ~115 | ND | ND | ND | ND | •Superoxide-generated post-acute EtOH exposure contributes to liver injury [67] •Cross-tolerance between EtOH intoxication and endotoxemia [68] |
Lieber-DeCarli, (L/D) diet | Ad lib feeding | 10 w–9 m | Mouse Rat | AST: ~120 | + | − | −/+ | − | •Convenient |
Two-hit model: ad lib L/D diet + LPS i.v./i.p. (0.5–1 ug/g) | 10 w | Rat | ALT: ~400 AST: ~ 500 | ++ | + | + | − | HSC activation [72] | |
Two-hit model: ad lib L/D diet + CCl4 (1 μl/g) | 3–5 w | Mouse Rat | ALT: ~350 AST: ~340 | + | −/+ | + | −/+ | ||
10-day ad lib L/D + single EtOH binge (5 g/kg) | 10 day | Mouse | ALT: ~250 AST: ~400 | + | − | + | − | •Mild and transient neutrophil infiltration [53] | |
Western ethanol diet plus weekly binge | Ad lib feeding of liquid diet high in cholesterol and saturated fat plus weekly ethanol binge (3.5–4.5 g/kg) | 8 w | Mouse | ALT: ~100 | +++ | ++ | +++ | +++ | •Easy procedure •Control mice also develop mild fatty liver •Activated hepatic stellate cells from the model have been characterized [75] |
Chronic intragastric infusion (iG) | Standard iG: EtOH (8–12 g/kg/day) | 10–16 w | Rat | ALT: ~460 AST: ~446 | ++ | + | ++ | + | •Sustained high blood alcohol levels |
Standard iG: high dose EtOH (27–32 g/kg/day) | 7–8 w | Mouse | ALT: ~250 | +++ | + | ++ | + | •Show EtOH dose-dependent liver injury [47] | |
Two-hit model: iG + enteral LPS (5 mg/kg, weekly) | 9 w | Rat | ALT: ~227 | ++ | ++ | +++ | ++ | •Two-hit model •Enteral LPS challenge potentiate liver injury [76] | |
Two-hit model: iG + iron dextran (~75 mg/kg, s.c.) | 4–16 w | Mouse Rat | ALT: ~500 AST: ~290 | +++ | ++ | +++ | +++ | •Two-hit model | |
More advanced ALD iG models | Please refer to Table 5.3 for details | ||||||||
Acute and Subacute ALD Models
Administration of ethanol through oral gavage or intravenous injection for a short period has been used to evaluate acute (up to 72 h) and subacute (up to 7 days) effects of ethanol exposure. The simplest method for acute ethanol exposure is single binge (oral gavage) with 4–7 g/kg body weight of ethanol. Binge elicits physiologic and biochemical responses to acutely raised BACs including oxidant stress [67], lipolysis [78], endotoxemia [68, 79], and hepatic microcirculation response to transplantation [80] and acetaminophen [81]. Binge suppresses both intestinal fatty acid oxidation and triacylglycerol synthesis [82], which lead to epithelial damage. Binge also causes a swift increase in ethanol metabolism (SIAM) [83] and mitochondrial DNA (mtDNA) depletion [62, 63]. SIAM is associated with significant increase in hepatic oxygen consumption, altered glucose/lipid metabolism, activation of Kupffer cells, and pericentral hypoxia [83, 84], which are responsible for production of cytokines and prostaglandins and subsequent hepatocyte damage [85]. Acute single binge also depletes mtDNA by 51 % after 2 h of ethanol administration, and this effect is prevented by 4-methylpyrazole, an inhibitor of ethanol metabolism, and attenuated by melatonin [62, 63]. Thus, acute single binge causes SIAM in the liver, resulting in oxidative stress, mtDNA depletion, and subsequent early liver injury. It is noteworthy that binge drinking also increases serum endotoxin and bacterial DNA levels in healthy individuals, supporting the clinical relevance of the observations made in animals [86].
Although acute single binge causes mild and transient increase in levels of BACs, serum ALT, and LPS, it produces minimal pathological change in liver [60, 61]. When oral binge is given every 12 h or 24 for 3–4 days, there is little liver pathology observed [64, 66]. A significant increase in serum ALT and mild liver pathology are induced when these binge models are co-administrated with LPS [61] or binge is given in obese animals [65] (two-hit models). For example, acute single binge followed by LPS (5 mg/kg, i.v.) causes a surge of serum ALT levels up to 760 U/L and submassive necrosis and neutrophil infiltration [61].
In conclusion, although the acute and subacute ALD models are convenient and have no or minimal mortality rate, they do not produce classic features of human ALD. However, they are useful for mechanistic evaluation of ethanol action, early phase of liver injury, and ethanol’s priming or sensitizing effects on hepatic responses to the second hit.
Chronic ALD Models
Chronic Binge Model
Enomoto et al. demonstrated time-dependent liver injury in rats by daily oral gavage of ethanol (5 g/kg body weight) for 8 weeks [66]. Mild steatosis and mild ASH were observed at 4 weeks and 8 weeks after daily binge, respectively. The authors further demonstrated that sensitization of Kupffer cells to LPS contributed to ethanol-induced liver injury. However, they noted that repeated binge also result in endotoxin tolerance depending on the duration of the treatment [87]. This model is simple but daily binge for such a long period requires high skill of oral gavage, and only limited liver pathology is achievable by the method.
Ethanol in Drinking Water Model
Feeding rodents ethanol in drinking water is the simplest method. Rats with 40 % (v/v) ethanol in drinking water for up to 29 weeks developed mild to moderate ASH and some fibrotic changes in the liver [56]. However, mice given 20 % (v/v) ethanol in drinking water for 21 weeks did not have significant elevation of serum ALT and histology showed mild hepatic steatosis [57–59]. Mild liver pathology produced despite the prolonged treatments and the lack of nutritional control are major limitations of this method.
Chronic Lieber-DeCarli Liquid Diet (L/D) Model
Related posts:
 Nonalcoholic Fatty Liver Disease in Children
Diagnostic Considerations and Clinical End Points for Nonalcoholic Steatohepatitis
Hepatic and Extrahepatic Malignancies in NAFLD
Nonalcoholic Fatty Liver Disease in Children
Diagnostic Considerations and Clinical End Points for Nonalcoholic Steatohepatitis
Hepatic and Extrahepatic Malignancies in NAFLD
 Clinical Features, Disease Modifiers, and Natural History of Alcoholic Liver Disease
Clinical Features, Disease Modifiers, and Natural History of Alcoholic Liver Disease
 Pathogenesis of Alcoholic Liver Disease
Pathogenesis of Alcoholic Liver Disease
 Epidemiology and Risk Factors for Alcoholic Liver Disease
Epidemiology and Risk Factors for Alcoholic Liver Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


