Fig. 3.1
Effects of heavy alcohol use on metabolic and signaling pathways leading to increased hepatic fat synthesis. Heavy alcohol use increases hepatic triglyceride content via increased uptake of fatty acids from the circulation and increased de novo synthesis. The increase in de novo synthesis is mediated by activation of transcription factors which induce the synthesis of a battery of enzymes involved in the conversion of acetyl-CoA to fatty acids. Abbreviations: ADH alcohol dehydrogenase, ALDH2 aldehyde dehydrogenase 2, SREBP-1c steroid response element-binding protein-1c, ChREBP carbohydrate-response element-binding protein, ER endoplasmic reticulum, SIRT1 sirtuin 1, ASMase acidic sphingomyelinase, AMPK AMP-activated protein kinase, TNFα tumor necrosis factor-α, MAT1 methionine adenosyltransferase 1, MS methionine synthase, ACC acetyl-CoA carboxylase, FAS fatty acid synthase, SCD stearoyl-CoA desaturase, GSH reduced glutathione, ROS reactive oxygen species, RNS reactive nitrogen species
Alcohol induces ER stress through several mechanisms, including direct effects of acetaldehyde [66], oxidative stress [67], and elevated homocysteine. Homocysteine is elevated because alcohol interferes with the methionine cycle, whereby methionine is converted to S-adenosylmethionine (SAM) by methionine adenosyltransferase 1 (MAT1). SAM is converted to S-adenosylhomocysteine (SAH) by donating a methyl group to an accepting molecule, and SAH is hydrolyzed to homocysteine, which is converted to methionine by either methionine synthase (MS) or betaine-homocysteine methyltransferase (BHMT). Homocysteine is elevated in chronically ethanol-fed animals and in human alcoholics [68–71], and SAM is decreased, due to folate deficiency and to decreased activity of methionine synthase [72].
Mice fed homocysteine rapidly developed ER stress [67]; mice fed alcohol developed large increases in homocysteine; treatment with betaine, which acts as a methyl donor for BHMT, decreased levels of homocysteine and SREBP-1c [65]. In micropigs fed alcohol and a folate-deficient diet, SREBP-1c levels correlated with homocysteine levels [73], and this was reversed by administration of SAM, presumably through its inhibitory effects on ER stress [74]. Additionally, transgenic mice expressing human BHMT were resistant to alcohol-induced increases in homocysteine and hepatic steatosis [67].
SREBP-1c is also activated by acetaldehyde [63], as well as lipopolysaccharide (LPS) and TNFα, all of which are increased in ALD [75] even at very early stages. In fact binge drinking by normal volunteers increased circulating LPS [76], and acute exposure to ethanol increases SREBP-1c mRNA and activity [77]. Inhibition of AMPK, discussed below, increased stability and activity of SREBP-1c [78]. Thus, many effects of alcohol converge to activate SREBP-1c (Fig. 3.1).
Carbohydrate-Responsive Element-Binding Protein
Carbohydrate -responsive element-binding protein (ChREBP) is a glucose-responsive transcription factor [79], whose activity is regulated by nuclear/cytosolic partitioning and posttranslational modification. ChREBP induces transcription of genes involved in glycolysis [liver pyruvate kinase (LPK)], lipogenesis (ACC and FAS), and gluconeogenesis (glucose-6-phosphatase (G6Pase) [80]. ChREBP is inhibited by phosphorylation by protein kinase A and AMPK [81]. AMPK activity is sensitive to the energy state of the cell; under low energy conditions, AMPK is active, and ChREBP is retained in the cytosol. When glucose is abundant, AMPK is suppressed, leading to nuclear translocation of ChREBP and induction of glucose-responsive genes. ChREBP activity is activated by dephosphorylation. Specific isoforms of protein phosphatase 2A (PP2A) are activated by xylulose-5-phosphate (Xu-5-P), an intermediate in the pentose phosphate shunt. When glucose is high, it is converted to Xu-5-P, activating PP2A and ChREBP [82].
Alcohol infusion significantly increased hepatic mRNA and protein expression of ChREBP in rats [83]. Treatment of hepatoma cells with alcohol activated a ChREBP-dependent reporter, an effect blocked by ChREBP siRNA. Alcohol caused nuclear translocation of ChREBP and increased its ability to bind DNA, leading to an increase in the expression of the ChREBP gene battery [84]. Alcohol may activate ChREBP via activation of PP2A and inhibition of AMPK, but not by an increase in hepatic Xu-5-P (see below and Fig. 3.1). Induction of SREBP and ChREBP by alcohol synergistically increases lipogenesis.
Liver X Receptor (LXR) and Farnesoid X Receptor
LXR is an orphan nuclear receptor [85] activated by glucose, glucose-6-phosphate, and oxysterols that is involved in cholesterol homeostasis [86–89]. LXR regulates lipogenic genes such as FAS and ACC, as well as SREBP-1c and ChREBP [90–92]. LXR agonists induced CD36, the liver fatty acid uptake transporter, and an interaction between PPARα and SREBP-1 may be mediated by LXR [93]. Farnesoid X receptor (FXR) is the bile salt receptor which is the target of agonist drugs in clinical trials for nonalcoholic fatty liver and cholestatic liver diseases. FXR activity is reduced in alcohol-fed mice [94]. FXR agonists reversed the inhibition of FXR activity, suppressed induction of CYP2E1, reduced oxidative stress, reduced SREBP-1c activity, and reduced steatosis in animal models of ALD [95].
Ceramide Signaling and the Role of Acidic Sphingomyelinase (ASMase)
Activation of SREBP-1c and ChREBP may be coordinated by the signaling lipid ceramide. Ceramide is generated either de novo from serine and palmitoyl-CoA, via serine palmitoyltransferase (SPT), or from the breakdown of sphingomyelin (mediated by sphingomyelinases: acidic sphingomyelinase (ASMase) and neutral sphingomyelinase (NSMase)). Ceramide is further metabolized by ceramidase to sphingosine, which can be converted back to ceramide by (dihydro)ceramide synthase. Ceramide levels are increased by alcohol (see below). Most recent interest has centered on the ASMase pathway. ASMase is activated by reactive oxygen species and by TNFα, and ASMase mRNA is increased in biopsies from individuals suffering from alcoholic hepatitis [96]. When ASMase activity is reduced (by genetic knockout or by use of inhibitors), alcohol-induced lipogenesis is reduced; there is less cholesterol accumulation in the mitochondrion, less steatosis, and reduced sensitivity to LPS [96]. In ASMase knockout mice, alcohol does not induce ER stress, despite elevated levels of homocysteine, and homocysteine administration no longer induced ER stress. Other effects of ceramide deduced from experiments with ASMase knockout animals include reduction in MAT1A mRNA and activity; inhibition of liver CTP-phosphocholine cytidylyltransferase (CCT), which participates in phosphatidylcholine synthesis; and activation of ER stress triggered by inhibition of autophagy [96]. As described below, ceramide may have effects on AMPK as well. Thus, strategies aimed at blocking ASMase activity and reducing ceramide levels seem like a promising direction of therapeutic research (Fig. 3.1).
Role of Endogenous Cannabinoids
The endocannabinoid system has been studied for its role in fatty liver diseases [97]. Liver cells express low levels of cannabinoid (CB1) receptors. Alcohol feeding increases the production of the endogenous cannabinoid 2-arachidonoylglycerol (2-AG) by HSCs, which could interact with hepatocytes in a paracrine fashion. Alcoholic steatosis was attenuated with CB1 antagonists, and CB1 knockout animals are resistant to alcoholic fatty liver [98]. In the knockout animals or animals treated with CB1 antagonists, the levels of SREBP-1c and FAS were reduced relative to control alcohol-fed animals, while CPT1 activity is increased. Activation of CB1 receptors increased the ability of LXR to induce SREBP-1c, and blockade of CB1 receptors increased protein kinase A and AMPK activity, inhibiting LXR action [99]. Thus, endocannabinoids, and by extension use of marihuana, may aggravate ALD through induction of SREBP and inhibition of AMPK.
Hepatic Fatty Acid Oxidation
Decreased fatty acid oxidation can cause hepatic steatosis. Impaired fatty acid oxidation plays an important role in ALD and is mediated by actions of alcohol on energy sensors and transcription factors.
AMP-Activated Protein Kinase (AMPK)
AMPK is a serine/threonine kinase which regulates energy homeostasis by stimulating energy-producing processes, e.g., fatty acid oxidation and glycolysis, and inhibiting energy-utilizing processes, e.g., fatty acid and amino acid synthesis. This occurs, in part, through phosphorylation and inactivation of ACC, decreasing the levels of malonyl-CoA, a precursor in the synthesis of fatty acids and an inhibitor of carnitine palmitoyltransferase 1 (CPT1). CPT1 is the rate-limiting enzyme in fatty acid oxidation. Thus, AMPK is able to exert its effects on both fatty acid synthesis and oxidation through its effects on ACC [100–102]. AMPK also inhibits SREBP-1c and ChREBP activity [78, 81, 103], reducing de novo lipogenesis.
AMPK activity is decreased in alcohol-treated hepatoma cells and in the liver, with a concomitant increase in ACC activity and steatosis [78, 104]. Activation of AMPK with metformin blocks the effect of alcohol on ACC activity, SREBP-1c, and reduces steatosis [78]. Control of AMPK activity is mediated by phosphorylation on a threonine residue at position 172; three main mechanisms for the effect of alcohol on AMPK have been reported (Fig. 3.2).
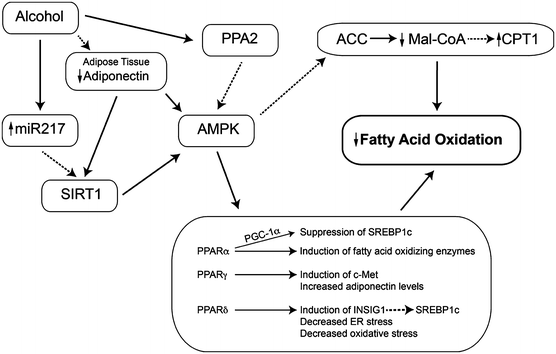
Fig. 3.2
Effects of heavy alcohol use on the controls of fatty acid oxidation. Heavy alcohol use reduces the rate of fatty acid oxidation in the liver. This is mediated in particular by inhibition of AMP-activated protein kinase (AMPK) (which modifies both the activity of SREBP-1c and the entry of fatty acyl-CoA esters into the mitochondrion) and peroxisome proliferator-activated receptors. As a result, fatty acids are stored as triglycerides. Abbreviations: SIRT1 sirtuin 1, AMPK AMP-activated protein kinase, PP2A protein phosphatase 2A, PPAR peroxisome proliferator-activated receptor, FFA free fatty acids, Mal-CoA malonyl-CoA, PGC-1 PPARγ coactivator-1, CPT1 carnitine palmitoyltransferase 1
Regulation of AMPK by PP2A and Ceramide
Protein phosphatase 2A (PP2A) is a serine/threonine phosphatase regulating cellular functions and signaling pathways, including ChREBP, apoptosis, and insulin signaling [105], and may be involved in alcohol inhibition of AMPK. A PP2A subunit co-immunoprecipitated with AMPK, alcohol increased PP2A activity in hepatoma cells [106], and the PP2A inhibitor okadaic acid or PP2A siRNA significantly attenuated the inhibitory effect of alcohol on AMPK phosphorylation.
PP2A is regulated by ceramide [107]; in fact, a form of PP2A was originally named the ceramide-activated protein phosphatase [108]. Alcohol treatment of hepatoma cells significantly increased cellular (C16 and C18) ceramide content and increased PP2A activity [106]. Inhibitors of the de novo synthetic pathway and NSMase did not prevent inhibition of AMPK by alcohol; however, the ASMase inhibitor imipramine did prevent this effect. This effect of alcohol was observed in vivo: in mice fed alcohol, there were 30 % increases in hepatic C16 and C18 ceramides, the level of ASMase mRNA was increased by 1.7-fold, and imipramine reduced ASMase activity, ceramide levels, PP2A activity, and triglyceride accumulation [109]. The increase in ASMase activity was not associated with increased levels of TNFα, and imipramine treatment did not restore AMPK activity. More recently, myriocin, an inhibitor of de novo ceramide synthesis, reversed alcoholic steatohepatitis in animals [110].
Alcohol-induced elevation of ceramide may contribute to the induction of steatosis by AMPK-dependent and AMPK-independent pathways.
Regulation of AMPK by Adiponectin
Adiponectin stimulates fatty acid oxidation, in part through its activation of PPARα and AMPK [38]. Alcohol feeding decreased plasma adiponectin levels [38, 47, 111] and the adiponectin receptors AdipoR2 and AdipoR1 in mice and micropigs, respectively [74, 111]. Treatment of alcohol-fed animals with adiponectin alleviated hepatic steatosis [58]. Conversely, adiponectin knockout mice are highly sensitive to alcohol-induced steatosis, but surprisingly no difference in AMPK activity or PPARα DNA-binding activity was observed [112]. Interestingly, adiponectin can reduce ceramide levels, suggesting another mechanism by which it improves liver metabolism in alcohol-fed animals.
Regulation of AMPK and Lipid Metabolism by SIRT1
SIRT1 is an NAD+-dependent protein deacetylase which senses the cellular redox state as a measure of energy need and can regulate the activity of proteins involved in lipid metabolism. SIRT1 is as a major regulator of AMPK [113]. Liver-specific knockout of SIRT1 results in hepatic steatosis and inflammation [114]. Alcohol feeding and in vitro studies show that metabolism of alcohol reduces SIRT1 activity [115, 116] and mRNA [117]. Mechanisms accounting for this effect include increased levels of miR217 in alcohol-metabolizing cells or liver [118]. This effect could be overcome with resveratrol, which also reversed steatosis [111], or feeding a high-saturated fat diet [115, 116]. Resveratrol increased circulating adiponectin and the activity of SIRT1, AMPK, and PPARγ coactivator-1α (PGC-1α) [111]. Reduction of SIRT1 activity has several downstream effects. Reduced SIRT1 activity leads to increased activity of SREBP-1c and changes the expression of lipin-1 (an Mg++-dependent phosphatidic acid phosphohydrolase). This protein is a regulator of fatty acid oxidation and de novo lipogenesis [119]. Alcohol feeding, via SIRT1 inhibition, increases the level of lipin1β, which in turn led to the activation of SREBP-1c. SIRT1 was also reduced in macrophages exposed to acetaldehyde or acetate, and they generated increased amounts of TNFα [120]. Feeding alcohol to SIRT1 knockout animals increased markers of inflammation, oxidative stress, ER stress, cytokine production, and fibrosis, suggesting an important role for SIRT1 in modulating all these effects of alcohol. In samples of liver from patients with alcoholic hepatitis, SIRT1 mRNA was reduced to about 25 % of normal, although the effects on lipin mRNA were more subtle [114]. The effect of alcohol on lipin expression was blocked by activators of AMPK or constitutively active AMPK genes. Thus, SIRT1 (and, by extension, dietary polyphenols such as resveratrol) plays important roles in regulating fat synthesis and oxidation in ALD.
Peroxisome Proliferator-Activated Receptors
Peroxisome proliferator-activated receptors (PPARs) are nuclear receptors that regulate genes involved in fatty acid oxidation and transport, lipid storage in adipocytes, and inflammation. Alcohol affects the activity of each of the three classes of PPARs.
Alcohol decreased the DNA-binding ability of PPARα, downregulating numerous PPAR-regulated genes [121]. Knockout of PPARα increased liver damage caused by alcohol feeding, including increased hepatomegaly [122]. PPARα inhibits the expression of SREBP-1c; hence, the reduction in PPARα activity by alcohol may be another mechanism of SREBP-1 induction. Pharmacological activation of PPARα prevents alcohol-induced fatty liver [123, 124]. Alcohol effects may involve AMPK, as the kinase may regulate PPARα through protein-protein interactions [125]. The action of PPARα depends on PGC-1α, which is activated by AMPK [126]. Alcohol feeding reduced the level of PGC-1α, and this was restored by changing the lipid in the diet to medium-chain triglyceride [117].
PPARγ is highly expressed in adipocytes, with low levels in the liver. It coordinates adipocyte differentiation, fatty acid uptake, and glucose metabolism. Based on pharmacological studies, PPARγ may play a role in ALD. Alcohol inhibited PPARγ transcriptional activity in cultured hepatocytes, but has also been shown to increase PPARγ mRNA [111, 127]. Treatment with the PPARγ agonist pioglitazone prevented the development of alcohol-induced steatosis [128]; however, pioglitazone also activates AMPK [129]. Pioglitazone was also reported to prevent alcoholic fatty liver through the upregulation of c-Met [130], an activator of lipid export (below).
PPARδ modulates energy metabolism and, like PPARα, inhibits SREBP-1c, possibly via induction of insulin-induced gene 1 (INSIG1), an inhibitor of SREBP-1c maturation [131]. PPARδ knockout animals had greater accumulation of triglyceride during alcohol feeding, increased SREBP-1c levels [132], and increased CYP2E1 mRNA. Furthermore, PPARδ agonists reduced alcohol-induced insulin resistance and oxidative stress [133], as well as ER stress and ceramide generation [134]. In a comprehensive comparison, activation of each class of PPARs ameliorated alcoholic steatosis [135].
The Role of Autophagy in the Regulation of Hepatic Lipid Stores
Autophagy is a process whereby intracellular organelles and membranes are engulfed in a particle called the autophagosome, which then fuses with lysosomes to form autolysosomes. This process prolongs cell survival during nutrient starvation: the breakdown of intracellular materials, including lipids, provides energy as a temporizing measure. A larger role for autophagy in lipid homeostasis is suggested by the finding that inhibition of autophagy leads to hepatic steatosis (as shown with the Atg7 knockout model). When autophagy is inhibited, the ER stress response is activated.
Studies on the effect of alcohol on autophagy show that acute alcohol gavage and chronic alcohol feeding increase the number of autophagosomes (estimated from the expression of the LC3II protein which is associated with these organelles). This appears to be dependent on alcohol metabolism and probably oxidative stress. It results from the inhibition of mTOR activity – the exact mechanism of this effect is unknown and is somewhat unexpected given that AMPK activation also leads to reduced mTOR activity and AMPK is decreased with alcohol feeding. A separate pathway regulating mTOR may thus play a role. It is not clear whether degradation of the contents of the autophagosome is altered in alcohol-fed animals: inhibition of fusion with lysosomes could contribute to the increase in autophagosomes. Autophagy may be a protective response to alcohol exposure, as suppression of autophagy made experimental alcoholic liver injury worse [136]. Autophagy may also serve to remove mitochondria damaged by heavy alcohol exposure [12]. The current state of this research, and the differences between acute and chronic exposure to alcohol, was recently reviewed [137].
Export of Triglycerides as Very Low-Density Lipoprotein
The liver packages triglycerides (TGs) into very low-density lipoprotein (VLDL) particles through the fusion of lipid droplets with apolipoprotein B100 (apoB100), which is dependent on the activity of microsomal triglyceride transfer protein (MTP). VLDL export is impaired in ALD through several mechanisms. Phosphatidylcholine (PC) is required for VLDL secretion, and the hepatic levels of this lipid are decreased with alcohol feeding. Two pathways produce PC: the majority is produced from choline, first phosphorylated and then converted to CDP-choline by CTP-phosphocholine cytidylyltransferase (CCT). CDP-choline then forms phosphatidylcholine by condensation with diacylglycerol. CCT is inhibited in alcohol-fed liver. Phosphatidylethanolamine methyltransferase (PEMT) produces perhaps 20–30 % of the needed PC by methylating phosphatidylethanolamine [138]. Inhibition of PEMT activity decreases PC levels and leads to steatosis [138, 139]. Since PEMT is a SAM-dependent enzyme, the low SAM levels in ALD leads to reduced PEMT activity [140, 141]. BHMT, which is decreased in ALD, can increase the expression of apoB100, probably via decreased levels of homocysteine [65, 142]. PPARα regulates the production of MTP; MTP levels were decreased in livers of alcohol-fed animals, while treatment with a PPARα agonist upregulated MTP and increased export of VLDL [123, 143, 144]. Hepatocyte growth factor (HGF) , acting though its receptor, encoded by the c-Met oncogene, stimulates the synthesis of apoB100 and MTP; although HGF levels are increased in alcohol-fed liver, c-Met levels are decreased [144]. Pharmacological administration of HGF overcomes this reduction in receptor level and reverses alcoholic steatosis [145]. Thus, impaired TG export contributes to ALD (Fig. 3.3). The potential mechanisms of steatosis in ALD are summarized in Table 3.1.
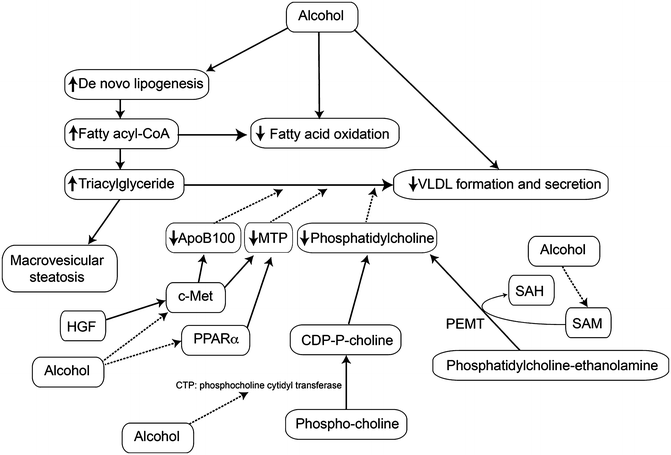
Fig. 3.3
Effects of heavy alcohol use on the export of triglyceride as VLDL. Heavy alcohol use increases the synthesis of fatty acids and triglycerides in the liver which are packaged as VLDL for export; however, alcohol use impairs this process below the rate needed to export all the excess triglyceride so that there may be both elevated VLDL in the circulation (hypertriglyceridemia) and hepatic steatosis. Abbreviations: VLDL very low-density lipoprotein, HGF hepatocyte growth factor, c-Met receptor for hepatocyte growth factor, MTP microsomal triglyceride transfer protein, apoB100 apolipoprotein B100, SAM S-adenosylmethionine, SAH S-adenosylhomocysteine, PEMT phosphatidylethanolamine methyltransferase
Table 3.1
Mechanisms by which alcohol use causes steatosis
Pathways | Mechanism | Consequences |
|---|---|---|
Increased FFA flux from adipose tissue | Reduced insulin sensitivity of adipose; adipose tissue inflammation | Elevated FFA delivered to the liver for storage |
Reduced adiponectin | Direct effect of alcohol on adipose | Reduced AMPK activity, reduced FFA oxidation |
Inhibition of methionine cycle | Inhibition of MAT1, MS: decreased SAM, increased homocysteine | ER stress, reduction in phosphatidylcholine synthesis and other methylation reactions |
ER stress | Oxidative stress, increased ceramide, increased homocysteine | Proapoptotic, increases SREBP-1c |
Altered miRNA levels | Unknown | Decreased SIRT1, increased intestinal permeability, sensitization of Kupffer cells |
Increased ceramide | Activation of ASMase by TNF, ER stress | Activation of PP2A, inhibition of phosphocholine synthesis, inhibition of AMPK, inhibition of MAT1 |
Inhibition of AMPK | Reduced SIRT1, increased ceramide, increased PP2A | Reduced fatty acid oxidation, reduced de novo fat synthesis |
Increased SREBP-1c activity | Reduced AMPK, reduced SIRT1, ER stress | Increased de novo fat synthesis |
Increased ChREBP activity | Decreased AMPK, increased PP2A | Increased de novo fat synthesis |
Reduced c-Met expression | Increased ceramide | Reduced MTP and apoB100 expression, decreased VLDL export |
Increased endocannabinoids | Increased SREBP-1c | Increased de novo fat synthesis |
Decreased PPAR activity | Acetaldehyde, decreased AMPK activity | Increased SREBP-1c, decreased c-Met, decreased fatty acid oxidation |
Inflammation and Cell Death in ALD
Fatty liver alone does not contribute to morbidity in alcoholic patients; as in nonalcoholic fatty liver, the development of inflammation, cell death, and ultimately fibrosis are much more important. Oxidant stress, endotoxemia, innate immunity, and apoptosis contribute to these processes.
Inflammation
Alcohol exposure impacts the inflammatory/immune response via a myriad of mechanisms and can either tolerize or sensitize the liver to a second inflammatory stimulus [146]. The effect of alcohol may be determined by the relative timing of the two responses. For example, if lipopolysaccharide (LPS) is administered during acute alcohol intoxication, the alcohol exposure blunts hepatic inflammation and injury caused by the LPS; in contrast, if LPS is given ~1 day after alcohol exposure or after chronic alcohol exposure, the inflammatory response is synergized. In general, however, it can be said that alcohol exposure enhances normal inflammatory tissue damage caused predominantly by the innate immune response.
Alcohol-Induced Endotoxemia
The strategic location of the liver between the intestinal tract and the rest of the body makes it a critical organ for clearance of xenobiotics and toxins that enter the portal blood. It is therefore not surprising that the liver has a very high capacity for phase I and II metabolic processes, which is responsible for the well-known “first-pass effect” in xenobiotic metabolism. As early as 1893, Pavlov observed that low levels of intestinally derived “toxins” are normally present in the portal blood and are cleared by the liver [147]. Kupffer cells, the resident hepatic macrophages, are responsible for clearing endotoxin [148] (Fig. 3.4). When high levels of LPS are present in the portal blood, Kupffer cells are activated and can mediate toxic responses in the liver [149]. It has been known since the 1970s that alcohol consumption increases the amount of detectable LPS in the blood [150, 151]. These increases are thought to be caused by both dysbiosis in the GI tract microbiome and a decrease in GI tract barrier function. Importantly, these are highly interdependent functions in that dysbiosis decreases GI tract barrier and altered barrier function can impact the microbiome.
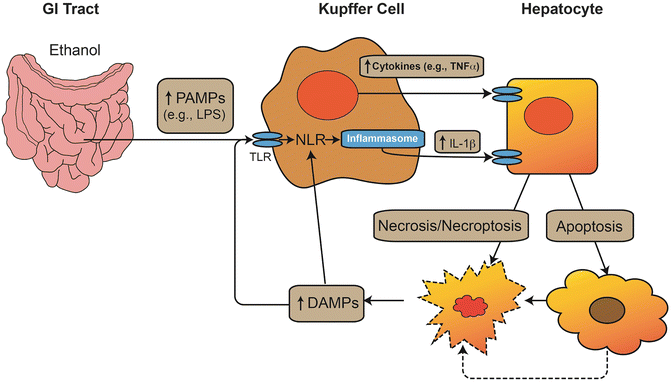
Fig. 3.4
Interaction between danger signals and the innate immune response in ALD. Alcohol increases gut permeability and increases the level of pathogen-associated molecular pattern molecules (PAMPs), such as lipopolysaccharide (LPS) in the portal blood. These PAMPs interact with toll-like receptors (TLR) to increase de novo synthesis of proinflammatory cytokines (e.g., TNFα). These signals also activate the inflammasome complex via NOD-like receptors (NLRs) to proteolytically activate IL-1β. These cytokines induce cell death via necrosis or apoptosis; furthermore, the energetic stress on the cells caused by alcohol exposure can lead to necrosis secondary to the initiation of apoptosis (i.e., necroptosis). Products of hepatocyte stress and death can serve as damage-associated molecular pattern (DAMP) molecules, which can also stimulate macrophages, either via TLR signaling or via direct activation of NLRs and the inflammasome. The result is a vicious cycle of inflammation and cellular injury. Alcohol enhances the inflammatory response of innate immune cells (i.e., priming effect) and exacerbates the cytotoxic effect of cytokines on parenchymal cells (i.e., sensitizing effect)
Dysbiosis
The concept that the microbiome can contribute to liver disease was first shown experimentally more than 60 years ago when nonabsorbable antibiotics protected against choline-deficiency-induced liver injury [152]. Today, the gut-liver axis, and enteric dysbiosis, is a generally accepted component in the development of liver disease. Indeed, ALD was a pioneering disease in which dysbiosis was considered a mechanism of tissue injury and commensal bacterial supplementation was demonstrated to protect against experimental ALD over 20 years ago [153].
The advent of platform-based analysis approaches (e.g., deep sequencing) and bioinformatics approaches has dramatically increased our understanding of how rapidly and dynamically the microbiome can change and affect change in ALD [154, 155]. These studies have validated early studies that indicated that alcohol exposure expands the population of Gram-negative bacteria in the gut, which explains, in part, the low-grade endotoxemia caused by alcohol. However, these studies, coupled with functional analyses, such as metabolomic profiling, have identified a much more active role of the microbiome in maintaining liver health. Indeed, recent work indicates that protective effects mediated by diet (e.g., saturated fat [156]) or by knocking out key genes (e.g., TNFR1 [157]) may have as much to do with the response of the microbiome to these changes as to the host’s [158, 159]. This rapidly advancing field will likely continue to yield new insight into ALD for the foreseeable future.
Decreased Gut Barrier Function
The ability of the mucosal barrier to withhold macromolecules is imperfect, and small amounts of antigens and other products normally traverse the intestinal wall. To a certain extent, this “leakiness” of the GI tract is important for both new antigen presentation and development of immune tolerance. However, if the barrier is significantly impaired, the innate immune response can be activated and can thereby induce a local and/or systemic inflammatory response. Alcohol has been known to increase the permeability of the GI tract to small molecules [160]. The barrier function of the GI tract is multilayered and includes physical barriers, such as the epithelial cells and the apical junctional proteins, as well as an active and robust immune response. Alcohol appears to decrease the barrier function via several of these pathways. Alcohol exposure directly damages epithelial cells and may cause GI tract erosions [160]. Alcohol also decreases the surface expression of apical junctional proteins (e.g., ZO-1), which are critical for maintaining normal tight junction barriers [161, 162]. Alcohol also induces a localized inflammatory response that again likely contributes to GI tract damage and decreased barrier function [155, 163]. Whereas the totality of the effect of ethanol on GI tract barrier function is incompletely understood, it is now well established that the barrier function is impaired and that the increase in permeability likely contributes to the initiation and development of ALD.
Priming of the Innate Immune System in ALD
The concept of priming and sensitization is important in ALD. Activation of the inflammatory response is, at least in part, due to increased levels of LPS [164, 165]. However, elevated levels of LPS found in alcoholics and in experimental ALD are relatively low compared to those found in experimental endotoxemia and human sepsis. Furthermore, liver injury caused by alcohol cannot be mimicked by chronic low-dose LPS in the absence of ethanol [166]. This is explained by the fact that inflammatory cells are primed to activation by alcohol administration, e.g., peripheral blood monocytes from patients with alcoholic hepatitis spontaneously produce proinflammatory mediators (e.g., TNFα). In response to LPS, these monocytes produce more proinflammatory mediators than their control counterparts [167]. Hepatocytes also appear to be sensitized to inflammatory stimuli by alcohol administration. Although TNFα is proproliferative in hepatocytes isolated from naïve animals, it is proapoptotic in cells isolated from alcohol-treated animals [168], via cellular “death domain” pathways [169]. HSCs also are sensitized by alcohol administration [170, 171]. The concept of priming and sensitization also means that there may be a series of sequential events in the progression of liver damage due to alcohol exposure. Specifically, the priming of inflammatory cells by alcohol leads to greater cell injury of sensitized hepatocytes; hence, blocking the activation of Kupffer cells [172] or employing knockouts with impaired Kupffer cell responses [173] prevents hepatocyte damage.
The enhanced production of TNFα is considered a key factor in the priming effect of alcohol on macrophage activation by LPS. Alcohol causes changes in the signaling cascade from the LPS receptor partners (CD14/TLR4) to the expression of TNFα. The expression of CD14 on Kupffer cells is upregulated by oxidant-dependent activation of the transcription factor AP-1 by alcohol [174]. Two transcription factors that are critical in TNFα expression are NFκB and EGR-1. Alcohol increases NFκB activation via both ROS-dependent [175] and ROS-independent pathways [176]. Ethanol also increases EGR-1 transcriptional activity via enhancing ERK1/ERK2 signaling [177]. Furthermore, alcohol itself enhances LPS-stimulated ROS production by macrophages via MyD88-independent TLR4 signaling [178]. Alcohol-induced TNFα expression is also regulated by increased mRNA stability. LPS stimulation of p38 mitogen-activated protein kinase contributes to this stabilization of TNFα mRNA. Moreover, studies have shown that at least one mRNA-binding protein, HuR, is also involved in the stabilization of TNFα mRNA stability after chronic exposure to ethanol (see [179] for review). The net effect is an increase in TNFα protein release in alcohol-exposed macrophages after stimulation.
The sensitization effect of alcohol on hepatocytes represents an increased sensitivity of parenchymal cells to cytotoxic killing. TNFα-induced signaling via TNFR1 appears to play a key role. In normal hepatocytes, TNFα is a mitogen; however, alcohol causes changes in intracellular signaling that switch this mitogenic response to a cytotoxic response. There is a critical role of CYP2E1 induction in this process [180]. Using the model of CYP2E1 induction by pyrazole, they have shown that sensitization was reduced by half in CYP2E1 knockout mice. Sensitization was associated with the activation of p38 and JNK kinases, and liver injury was blocked with inhibitors of these kinases. Liver mitochondria from animals treated with pyrazole and TNFα underwent increased swelling in response to calcium, and cyclosporine A, an inhibitor of the mitochondrial permeability transition, reduced liver injury. Pyrazole increased the levels of IkB, which reduces the activation of NFκB by TNFα. NFκB is thought to mediate cell-protective effects of TNFα; this inhibition would tilt the effect of TNFα toward cell injury. The sustained activation of JNK (which is an apoptotic signal) coupled with the inhibition of NFκB activation are proposed to mediate the sensitization effect of alcohol (see [181] for review). Administration of N-acetylcysteine to mice treated with pyrazole and LPS or TNFα ameliorated in the changes in p38, JNK, and activation of NFκB. Further, pyrazole plus TNFα administration caused less liver injury in NOS2 (iNOS) knockout animals than in controls, indicating that peroxynitrite radicals contributed to sensitization of the liver. Thus, there is strong evidence from animal studies that oxidative stress, in particular that resulting from induction of CYP2E1, is very important in response of the liver to increased levels of LPS and TNFα.
MicroRNA in ALD
MicroRNAs are ubiquitous regulators of gene expression, which interact with mRNAs and can either block translation or stabilize the mRNA. Levels of a host of miRNAs have been reported to be altered in alcohol-fed animals or in cultured cells (reviewed by McDaniel et al. [182]). Some miRNAs are increased (miR21, miR34a, miR155, and miR320) and some are decreased (miR122, miR181a, miR199a, and miR200a) by alcohol exposure. Recent studies have indicated that these miRNAs are likely involved in many of the pathological responses to alcohol already discussed, including the modulation of SIRT1 (miR34a) and of intestinal permeability (miR122), apoptosis [183], and the sensitization of Kupffer cells to LPS (miR155). These RNAs may be targetable for therapy of ALD and may also provide serum biomarkers for different stages of the disease.
Contribution of Cell Death to Inflammation
As mentioned above, the strategic location of the liver between the intestinal tract and the rest of the body makes it a critical organ for clearance of xenobiotics and toxins that enter the portal blood; as such, the liver has a high likelihood of toxic injury. Hepatocellular death, when controlled, may clear damaged or dysplastic cells and thereby protect the organism as a whole; however, uncontrolled or high levels of cell death can induce toxic inflammatory responses.
Apoptosis, Necrosis, and Necroptosis
The two major death responses available to the cell are necrosis and apoptosis (Fig. 3.4). Necrosis is generally an acute response to traumatic stress and is characterized by frank autolysis of the cell and, in its most severe form, is independent of any intracellular signaling process. Apoptosis is a controlled ATP-dependent response to intrinsic and/or extrinsic stimuli. In contrast to necrosis, cellular fragments are released as blebs, and there is no release of intracellular contents into the extracellular space. As will be discussed below, necrosis is considered more proinflammatory by inducing the inflammasome. Both apoptosis and necrosis are validated histologies in fatty liver diseases, including ALD. However, the relative contribution of apoptosis histologically to ALD is lower than would be expected by in vitro studies or in studies with inhibitors of apoptosis (e.g., [184]). It is now understood that apoptosis and necrosis are not distinct events, but rather part of a continuum of cellular responses to cytopathic stress. In this, continuum may extend to very early effects of alcohol, such as ER stress. ER stress was reported to stimulate the association of IRF3 with an adaptor protein (stimulator of interferon genes or STING) and the activation of IRF3, which is proapoptotic. This effect was independent of the development of inflammation [185]. An apoptotic response may convert to necrosis, if cellular energy stores are insufficient to complete the apoptotic process (i.e., “necroptosis”) [186]. The metabolic stress caused by alcohol exposure that is described above (Sections “Metabolism of Ethanol and Its Role in Oxidative Stress” and “Effects of Alcohol Consumption on Liver Fat Homeostasis”) may thereby cause necroptosis [187].
Stimulation of the Inflammasome by Cell Death
Inflammation is induced not only by pathogen-associated molecular patterns (“PAMPs”), such as LPS (see section “Contribution of cell death to inflammation”), but also by molecules released from dead or dying cells (damage-associated molecular patterns (DAMPs)) (Fig. 3.4). These danger signals interact with the toll-like receptor (TLR) family of pattern recognition receptors, which thereby activate intracellular sensor molecules, the NOD-like receptors (NLRs), to subsequently activate caspases. The net result is the proteolytic activation and release of IL-1β, which induces the innate immune response [188]. As apoptosis does not release intracellular contents into the extracellular space, it is assumed to be less likely to induce the inflammasome response. However, it should be emphasized that apoptosis is not completely benign. Apoptotic bodies have been shown to activate the innate immune response, as well as to stimulate HSCs in the liver [189]. Another emerging area of intercellular communication in response to stress is the release of exosomes. Exosomes are small vesicles of extracellular membrane released by cells (e.g., hepatocytes) in response to stimuli. These exosomes can serve as DAMPs to stimulate the inflammasome response [190].
Plasminogen Activator Inhibitor-1 (PAI-1) and Its Potential Role in ALD
PAI-1 is an acute phase protein that is typically expressed in adipocytes and endothelial cells, but it can be induced in response to stress [191]. The classical role of PAI-1 is to inhibit the plasminogen activators, tPA and uPA, thus blocking activation of plasminogen to plasmin and fibrinolysis. A number of studies also implicate PAI-1 in ALD.
Acute and chronic alcohol-induced steatosis is prevented by genetic or pharmacologic inhibition of PAI-1 expression [192]. Plasminogen activators cleave latent hepatocyte growth factor (HGF) to the mature active form [193, 194], which is important in lipoprotein secretion [195] (see section “Export of Triglycerides as Very Low-Density Lipoprotein”). Indeed, genetic or pharmacologic inhibition of PAI-1 expression caused an increase in c-Met phosphorylation, apoB100 expression, as well as VLDL secretion after alcohol exposure [192]. These data suggest that the protective effect of PAI-1 inhibition is due to a compensatory increase in VLDL synthesis.
In addition to blocking steatosis, preventing PAI-1 induction completely protected against chronic alcohol-induced inflammation [192]. The “classical” role of PAI-1 in impairing fibrinolysis may contribute to inflammation (see below). Furthermore, PAI-1 may alter the profile of other inflammatory mediators via inhibition of plasminogen activators independent of fibrin. For example, the inhibition of plasmin activation by PAI-1 prevents the conversion of secreted latent TGFβ to its active form [196], which may mediate anti-inflammatory effects, especially on monocytes/macrophages [197]. These potential mechanisms are not mutually exclusive and may occur in tandem [198].
Extracellular Matrix (ECM) Changes During ALD
The role of the ECM is well established in the setting of chronic liver injury, such as ALD. Indeed, robust changes in the hepatic ECM are the hallmark of later stages of liver disease (fibrosis and cirrhosis) which are characterized by dramatic increases in collagen deposition [199]. Given the dominance of collagenous changes in the hepatic ECM during fibrosis/cirrhosis, many studies and previous reviews have focused on the underlying changes in collagen deposition.
Transitional Extracellular Matrix Changes and Inflammatory Liver Damage
Hepatic ECM changes in response to alcohol are not limited to fibrogenesis per se. Indeed, it is becoming increasingly understood that the hepatic ECM responds dynamically to stress (Fig. 3.5). For example, changes in the expression of ECM proteins such as complement and fibrin (see below) and fibronectin [200, 201] have been observed in models of hepatic inflammation. Importantly, blocking these ECM changes blunts, at least in part, hepatic injury in these models. Other alterations of the ECM include changes in the deposition and distribution of laminin and vitronectin [202]. These dynamic changes in the matrix likely play key roles in mediating and coordinating the (patho)physiologic responses of the tissue.
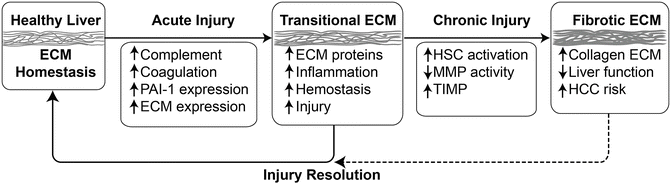
Fig. 3.5
Matrix alterations in response to acute and chronic damage in the liver. Although remodeling in response to chronic injury (i.e., fibrosis) is well known in the context of chronic liver injury, the hepatic extracellular matrix (ECM) also responses dynamically to acute stress. Key events in response to acute stress include activation of the complement and coagulation cascades, as well as induction of key genes (e.g., PAI-1 and osteopontin) that directly or indirectly mediate qualitative and quantitative changes to the ECM. These acute responses can be viewed as an arm of the wound-healing response and facilitate recovery from damage, which resolves once the damage is repaired. However, under conditions of chronic injury, these changes contribute to activation of a significant remodeling response that leads to scar formation. This latter response is known as fibrogenesis in the liver. Other abbreviations: MMP matrix metalloproteases, TIMP tissue inhibitors of metalloproteases, PAI-1 plasminogen activator inhibitor-1, HCC hepatocellular carcinoma
PAI-1 and Hepatic Fibrosis
Plasmin degrades other extracellular matrix (ECM) proteins, such as laminin, proteoglycan, and type IV collagen [203–205] in addition to fibrin. Furthermore, plasmin activates MMPs, thereby indirectly degrading other ECM proteins [206]. Thus, by blocking plasminogen activation, PAI-1 could significantly alter the ECM. Indeed, a protective effect of pharmacologic/genetic prevention of PAI-1 induction has been observed in models of renal, pulmonary, and vascular remodeling, which share mechanisms with hepatic fibrosis [207–209]. The effect of knocking PAI-1 on hepatic fibrosis was first determined in a bile duct ligation model [210, 211]. In brief, PAI-1−/− mice were dramatically protected against ECM accumulation and fibrosis. A similar profibrotic effect of PAI-1 was observed in the AngII model of hepatic fibrosis [212], in which the protective effects of knocking out PAI-1 correlated with elevated degradation of ECM.
In contrast, liver damage and fibrosis were dramatically enhanced in PAI-1 knockout mice after chronic CCl4 exposure [213]. This may be secondary to an impaired hepatocyte proliferative response. A proproliferative role of PAI-1 was observed in mouse liver in experimental acetaminophen toxicity [214]. It is hypothesized that this impaired ability of the PAI-1 knockout liver to restitute itself enhances the amount of liver damage and accelerates the fibrotic process during chronic CCl4 exposure. The exact contribution of PAI-1 to human ALD awaits clinical trials of inhibitors of this protein, correlations of PAI-1 activity with liver injury (for instance, in alcoholic hepatitis), or implication of the PAI-1 locus in genome-wide association studies that are now under way.
The Complement Cascade and ALD
The complement system is a complex component of the innate immune system. It consists of soluble and membrane-bound proteins functioning by stepwise proteolytic activation. The complement cascade can be activated via three pathways: the classical, lectin, or alternative pathways. Activation of any of these pathways can lead to the cleavage of C3 and subsequent downstream activation of C5 and the rest of the terminal pathway. Hence, C3 plays a pivotal role in the complement system. In addition to producing complement proteins, cells in the liver also express complement factor receptors, as well as intrinsic regulatory proteins. Under basal conditions, Kupffer cells and HSCs express C3a and C5a receptors [215]. C5a receptor expression can be induced in proliferating hepatocytes or in response to inflammatory cytokines [215].
Recently, it has been suggested that complement activation contributes to the pathogenesis of various forms of liver diseases, including ALD [215]. In animal models, it was shown that chronic alcohol feeding increased activation of C3 [216], as well as increased accumulation of C3 or its proteolytic end product C3b/iC3b/C3c in the liver. Mice deficient in C3 and C5 are protected against alcohol-induced steatosis and increased transaminases [216]. In line with these findings, chronic alcoholic liver injury is exacerbated in mice lacking CD55/DAF, a complement regulatory protein that prevents the assembly of C3 convertase complexes and blocks the formation of the final membrane attack complex [216]. Moreover, alcohol feeding activates the classical complement pathway via C1q binding to apoptotic cells in the liver. Deletion of C1q prevents ethanol-induced complement activation, blunts TNFα and IL-6 expression in the liver, and reduces alcohol-induced liver damage [217]. In humans, the level of complement is increased in alcoholics during withdrawal, but is reduced in alcoholics with cirrhosis [218]. Therefore, future studies of complement in human ALD may help identify new therapeutic targets to treat this disease.
The Coagulation Cascade and Fibrin ECM in ALD
The liver is the major organ regulating the fibrin coagulation system. Fibrin metabolism is regulated via two pathways, coagulation and fibrinolysis [219]. Inhibition of fibrinolysis by plasminogen activator inhibitor-1 (PAI-1) can cause fibrin ECM to accumulate, even in the absence of enhanced fibrin deposition by the thrombin cascade [220]. Hepatic injury often involves dysregulation of the coagulation cascade and fibrinolysis, resulting in the formation of fibrin clots in the hepatic sinusoids [221, 222]. Fibrin clots block the blood flow within the hepatic parenchyma, causing microregional hypoxia and subsequent hepatocellular death [223, 224].
Fibrin ECM not only serves as a physical structure but also binds/interacts with several cellular biomolecules. One family of receptors that mediate these interactions is the integrins. Integrins transfer information from the ECM to the cell, allowing rapid and flexible responses to changes in the environment. Integrins play a myriad of roles within the body, including proliferation/angiogenesis, maintenance of differentiation, as well as inflammation and apoptosis [225, 226]. Integrins are found on almost all cell types in the liver. Therefore, altering the composition of the ECM has the potential to alter inflammatory signaling in the liver via a myriad of mechanisms.
Elevated PAI-1 levels and hypofibrinolysis are common during the development of ALD [227]. Indeed, PAI-1 levels during disease development are a predictor of later severity [228]. A recent human study further supports the hypothesis that PAI-1 plays a critical role in ALD [229]. However, few clinical studies have focused on the potential of inhibiting PAI-1 to slow the development of ALD. Indeed, it is well known that bleeding as well as thrombosis is common in end-stage liver disease [230], and there has been the slow realization that cirrhosis is a hypercoagulable state. In light of this, it is intriguing that anticoagulation with enoxaparin was reported to prevent decompensation in cirrhotics with portal vein thrombosis [231].
Osteopontin
Osteopontin, also designated secreted phosphoprotein-1 (SPP-1) , has been intensively studied for many years. It predominantly serves as an extracellular structural glycoprotein. Its synthesis is greatly upregulated in animal and human ALD by the increased exposure to LPS and has been linked to the activation of HSC and liver fibrosis (reviewed by Seth et al. [232, 233]) and with poor outcomes in alcoholic hepatitis [234]. However, very recent work with genetically modified mice has indicated that overexpression of SPP-1 prevents early ALD, in part by way of its ability to bind LPS [235], and knockout of SPP-1 promotes the neutrophilic infiltration of the liver in a model of alcoholic hepatitis [236]. The roles of osteopontin at various stages of ALD are thus open to further investigation, which will obviously bear on any therapeutic application of this protein.
Related posts:
 Nonalcoholic Fatty Liver Disease in Children
Diagnostic Considerations and Clinical End Points for Nonalcoholic Steatohepatitis
Hepatic and Extrahepatic Malignancies in NAFLD
Nonalcoholic Fatty Liver Disease in Children
Diagnostic Considerations and Clinical End Points for Nonalcoholic Steatohepatitis
Hepatic and Extrahepatic Malignancies in NAFLD
 Clinical Features, Disease Modifiers, and Natural History of Alcoholic Liver Disease
Clinical Features, Disease Modifiers, and Natural History of Alcoholic Liver Disease
 Animal Models of Alcoholic Liver Disease
Animal Models of Alcoholic Liver Disease
 Epidemiology and Risk Factors for Alcoholic Liver Disease
Epidemiology and Risk Factors for Alcoholic Liver Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


