Chapter 8A Pancreatic cancer and premalignant tumors
Molecular aspects
Pancreatic Cancer Overview
Pancreatic ductal adenocarcinoma (PDA), commonly known as pancreatic cancer, is the fourth leading cause of cancer death in the United States, preceded only by lung, colon, breast (in women), and prostate (in men) cancers (American Cancer Society, 2009). The diagnosis of PDA is devastating because it has one of the highest mortality rates of any cancer (American Cancer Society, 2009). To underscore this point, in 2009 approximately 42,470 Americans were diagnosed with PDA, and about 35,240 will die from it (American Cancer Society, 2009). A diagnosis of PDA is met with the daunting statistic that the patient has an overall 5-year survival rate of less than 5%.
The past 2 decades have seen an exponential increase in our understanding of the molecular basis and etiology behind PDA (Hruban et al, 2001; Jones et al, 2008, 2009). Still, the clinical management of this disease—including primary prevention, early detection, and better targeted treatment options—has not changed significantly over the past decade. Currently, the only cure for this disease is surgical resection. Unfortunately, only 20% of the patient population presents with resectable disease (Winter et al, 2002).
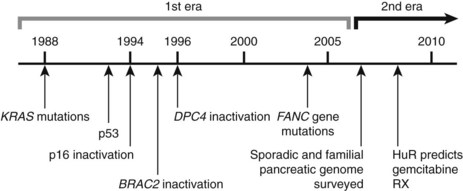
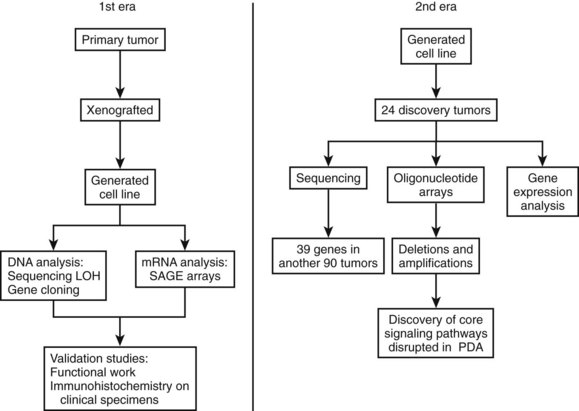
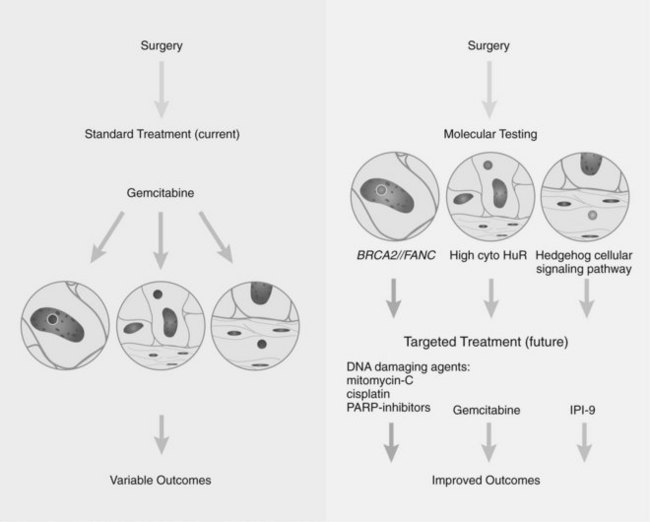
Research studying PDA can be simplistically divided into two eras (Fig. 8A.1). The first is highlighted by the discovery and development of technologies such as gene cloning and isolation of pure tumor tissue (i.e., xenografting primary tumors postresection; Fig. 8A.2, left). This era included landmark studies defining the genomic region of BRCA2 (Goggins et al, 1996), discovering SMAD4 (Hahn et al, 1996) as a tumor suppressor in PDA, activation of KRAS (Almoguera et al, 1988) as an early and frequent event in pancreatic tumorigenesis, and silencing of the CDKN2A (Caldas et al, 1994a) gene. As the result of advances in a number of molecular techniques, we are now upon the early stages of a new second era in genetic pancreatic cancer research, which combines high-throughput sequencing, gene expression and comparative genomic hybridization (CGH) arrays with statistical computational analysis, and large clinical databases (Fig. 8A.2, right) (Jones et al, 2008, 2009). This second era has already produced landmark publications that include a precise survey of multiple pancreatic cancer genomes both in sporadic and familial forms of the disease. This chapter aims to 1) provide a summary of the past two decades of pancreatic cancer research; 2) show that even though every tumor has a number of common molecular events, it is the differences between tumors that have clinical implications (Fig. 8A.3); and, finally, 3) set the stage for the future, which will include a discussion of successful early detection and treatment strategies for this deadly disease (Fig. 8A.3).
Progression Model of Pancreatic Ductal Adenocarcinoma
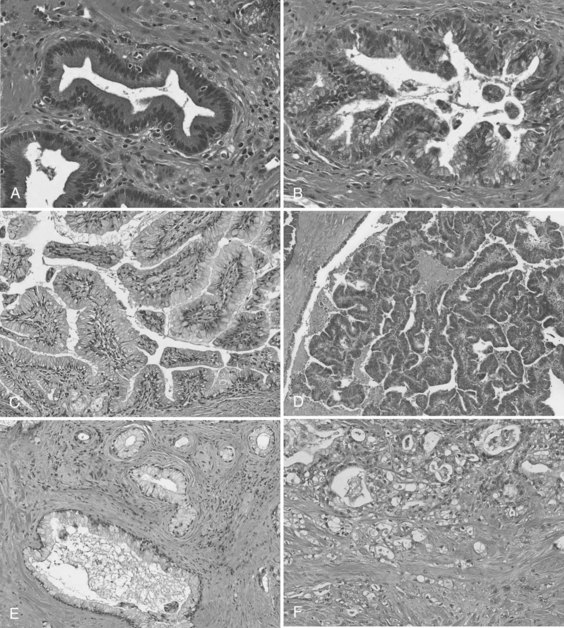
In the first era of pancreatic cancer research (see Figs. 8A.1 and 8A.2), the fields of molecular biology and pathology combined to establish a paradigm that PDA culminates from a multistep progression model (Hruban et al, 2001). This slow, sequential process may be the reason PDA is primarily a disease of people in their sixth and seventh decades of life. Definable pathologic markers on this stepwise progression—which follows a similar model first developed in colon carcinogenesis—are lesions referred to as pancreatic intraepithelial neoplasia (PanIN) (Hruban et al, 2001; Figs. 8A.4A and B). These lesions are believed to be precursor lesions to pancreatic cancer. PanIN lesions are thought to develop years before the emergence of PDA and are pathologically graded from low-grade PanIN lesions, PanIN-1 (Fig. 8A.4A), to high-grade lesions, PanIN-3 (see Fig. 8A.4B). Although low-grade PanIN lesions are common incidental findings, high-grade PanIN lesions are more common in pancreata with PDA.
Key evidence that PanIN lesions are precursors to PDA is that similar hallmark molecular defects are found in PanIN lesions adjacent to invasive cancers (Biankin et al, 2003; Luttges et al, 2001a; Maitra et al, 2002; van Heek et al, 2002; Wilentz et al, 2000a). However, although KRAS mutations are frequently found in early PanIN lesions (Shi et al, 2009), genes involved in DNA repair mechanisms, such as TP53 and BRCA2, are altered late in this progression model. Because these data are based on limited sample numbers and data points, it remains somewhat counterintuitive that an oncogenic stimulus or stimuli (e.g., KRAS activation) precedes DNA damage (mutations). Recently, it was shown that higher density PanIN lesions were found in familial PDA patients compared with patients with sporadic forms of PDA, again arguing in favor of the hypothesis that PanIN lesions are true precursors to PDA (Shi et al, 2009).15
Intraductal Papillary Mucinous Neoplasm
Intraductal papillary mucinous neoplasms (IPMNs) are a well-accepted clinical and pathologic entity (Fig. 8A.4C and D; see Chapter 57) (Hruban et al, 2004). IPMNs typically produce radiographically identifiable pancreatic ductal dilation, which may predominantly involve the main pancreatic ducts (main-duct type IPMN), the secondary ducts (branch-duct type IPMN), or both types of ducts (mixed type). The distinction between the branch-duct type and main-duct type is important, because the former arise in younger patients, are more likely to involve the head and uncinate process of the pancreas, and are associated with lower-grade dysplasia and fewer invasive carcinomas. Approximately 30% to 40% of resected IPMNs harbor an invasive adenocarcinoma, and adenocarcinoma is most strongly associated with main-duct IPMNs. Approximately half of invasive carcinomas arising within IPMNs are so-called colloid (mucinous) carcinomas, and most of the remainder are tubular adenocarcinomas, the latter being histologically indistinguishable from invasive ductal adenocarcinomas that arise in the setting of PanINs (Adsay et al, 2002). Colloid carcinomas associated with IPMNs have a relatively good prognosis compared with other pancreatic carcinomas of the ductal type, which have a 5-year survival of 60% (Maire et al, 2002).
PanINs and IPMNs show some overlapping features, such that both are inherently intraductal lesions composed predominantly of columnar, mucin-producing cells that may grow in a flat configuration or may produce papillae; these lesions show a range of cytologic and architectural atypia and can give rise to invasive adenocarcinomas of the pancreas (Fig. 8A.4E and F). An important feature that distinguishes the two lesions is that PanINs are microscopic lesions, and IPMNs are macroscopic. Nevertheless, recognition of an IPMN and its distinction from a PanIN lesion is important for two reasons: 1) IPMN-associated colloid carcinomas have a significantly better prognosis than either PanIN- or IPMN-associated tubular adenocarcinomas (Adsay et al, 2001), and 2) IPMNs have a propensity to be multifocal lesions, therefore patients who undergo partial pancreatectomy and are left with a remnant pancreas need to be followed for life, even when the lesion originally resected was a noninvasive IPMN (Sohn et al, 2004).20
Genetic analyses of IPMNs have disclosed abnormalities in many of the same genes altered in conventional ductal adenocarcinoma, including mutations in the KRAS2, TP53, and CDKN2A genes, although the frequency and stage of neoplastic progression at which these alterations occur in IPMNs differ from PanINs (Hruban et al, 2004; Sessa et al, 1994). For example, the Peutz–Jeghers gene STK11 is inactivated in one third of IPMNs, and some patients with Peutz–Jeghers syndrome develop IPMNs (Sahin et al, 2003); this is in contrast to conventional ductal adenocarcinomas, in which loss of STK11 is a rare event. In contrast to ductal adenocarcinomas and PanIN-3 lesions, abnormalities in the SMAD4 gene seem to be rare in IPMNs (Iacobuzio-Donahue et al, 2000).
Another distinction between PanINs and IPMNs relates to the expression of the caudal differentiation factor CDX2, a marker of intestinal differentiation. Most IPMNs express CDX2, in particular IPMNs associated with an invasive colloid carcinoma, whereas this is uncommon both in PanINs and in the subset of IPMNs that give rise to invasive cancers resembling ductal adenocarcinomas (Adsay et al, 2004). CDX2 expression in IPMNs usually is associated with expression of MUC2, an intestinal epithelial apomucin, whereas absence of CDX2 usually is associated with expression of MUC1, a biliary apomucin, and concomitant lack of expression of MUC2. These findings have suggested that there may be two divergent pathways of carcinogenesis within the pancreatic ducts (Adsay et al, 2002). The first is a so-called intestinal pathway that gives rise to CDX2– and MUC2-expressing IPMNs that progress to the better-prognosis colloid carcinomas. The second is a pancreatobiliary pathway that gives rise to CDX2-negative, MUC2-negative, MUC1-expressing PanINs and a subset of IPMNs, both of which can progress to the poorer-prognosis conventional ductal adenocarcinomas. More recently, it has been observed that IPMNs with moderate dysplasia had reduced or absent pp32 nuclear expression (Brody et al, 2007). This may be functionally significant, because ANP32A acts as a tumor suppressor in pancreatic and other cancers, and the absence of this protein may facilitate the tumorigenesis process.
Genetics of Pancreatic Ductal Adenocarcinoma
Genomic (DNA) Alterations in Pancreatic Cancer
The multitude of genetic abnormalities in pancreatic cancer have many characteristics similar to other solid tumors, thus they include point mutations in critical genes, chromosomal (copy number) aberrations, mitochondrial DNA mutations, telomeric abnormalities, and epigenetic silencing by methylation of defined promoter DNA sequences. Recently, it was estimated that an individual pancreatic tumor contains, on average, 63 genetic alterations, primarily point mutations. It is also believed that only a small subset of these mutations is required for tumorigenesis (Jones et al, 2008).
The field of analyzing the genetics of pancreatic cancer can be broken down chronologically (see Fig. 8A.1). First, landmark studies starting in the late 1980s and spanning nearly two decades are highlighted by the discovery of KRAS activation, SMAD4 and BRCA2 mutations, and CDKN2A silencing (see Fig. 8A.1 and Fig. 8A.2, left). Some of these discoveries not only opened up lines of investigation in the field of pancreatic cancer but also opened up avenues with other tumor types and specific classification of PDA subtypes (Iacobuzio-Donahue et al, 2009). Then, in 2008 and 2009, Jones and colleagues, with the help of advanced DNA sequencing technology, were able to perform high-throughput genetic analysis on various pancreatic cancer genomes (see Figs. 8A.1 and 8A.2, right).
Copy-Number Aberrations
Although now considered primitive, valuable cytogenetic analysis performed over a decade ago found that chromosomal aberrations occur in virtually every pancreatic cancer. Cytogenetic analyses of pancreatic cancers have shown multiple, nonrandom numerical and structural changes (Griffin et al, 1995; Sirivatanauksorn et al, 2001). The most common numerical abnormalities include losses of chromosomes 6, 12, 13, and 18 and gains of chromosomes 7 and 20. Structural abnormalities (intrachromosomal break points) frequently involve 1p and 1q, 3p, 4q, 6q, 7q, 17p, 11p, 11q, 15q, 16q, and 19q (Rigaud et al, 2000). The technical limitations of conventional cytogenetics has presented challenges for identifying genes that are affected by chromosomal breaks. Allelotyping identifies areas of gross chromosomal loss by using polymorphic microsatellite markers to determine regions of genomic loss compared with matched normal tissues, also known as loss of heterozygosity (LOH) analysis. Allelotyping operates on the basic principle of the two-hit hypothesis, which postulates that tumor suppressor genes require biallelic inactivation. This most commonly happens by intragenic mutation in one allele, followed by loss of genetic material in the other allele. Identifying regions of single or biallelic loss or mutation holds the potential to understand the role of neighboring novel and well-known tumor suppressor genes. A landmark allelotype analysis of pancreatic cancers was performed using approximately 80 pancreatic cancer xenografts and 386 microsatellite markers (Iacobuzio-Donahue et al, 2004). This work discovered allelic losses in chromosome regions in proximity to tumor suppressor genes CDKN2A, TP53, and SMAD4. Allelotype analysis of PanIN lesions also has been performed using microdissected samples, and as expected, LOH is seen in many of the same chromosomal regions as invasive cancer, including 9p, 17p, and 18q (Luttges et al, 2001b; Yamano et al, 2000). Although the changes are conserved in most synchronous precursor lesions (i.e., the same allele is lost in PanIN and associated cancers), there is occasional clonal divergence between high-grade PanIN lesions harboring genetically distinct changes from the synchronous invasive cancer (Yamano et al, 2000). These findings may have important clinical implications in regard to tumor heterogeneity and clonal cancer cell drug resistance.
CGH identifies genomic amplifications and deletions and differentially labels normal and tumor genomic sequences with different dyes. The relative ratio of the two dyes indicates regions of cancer-associated losses or gains, with a ratio of 1 : 1 consistent with no change in copy number compared with normal DNA. Conventional CGH is performed on metaphase spreads and suffers from low resolution and inability to precisely map the various regions of amplifications and deletions (Mahlamaki et al, 1997). The resolution of array CGH is significantly better than the conventional technique, ranging from 500 kb to 30 kb, permitting the precise mapping of deletion and amplicon boundaries and genes targeted therein. Array technology also provides the ability to more efficiently use probes to study amplification of a larger number of genes. Array CGH analysis of pancreatic cancers has identified numerous recurrent copy-number aberrations, including amplifications of c-MYC (8q), EGFR (7p), KRAS (12p), AKT2 (19q), and NCOA3 (20q) and deletions of SMAD4 (18q), CDKN2A (9p), FHIT (3p), and MAP2K4 (17p) (Aguirre et al, 2004; Calhoun et al, 2006; Holzmann et al, 2004).
Utilizing high-density single-nucleotide polymorphism arrays, Calhoun and colleagues surveyed all the commercially available pancreatic cancer cell lines. In brief, this study provided high resolution and detailed break-point mapping of these cell lines and found two subclasses of cancer cell lines, original chromosomal instability (CIN) and holey CIN genotypes (Calhoun et al, 2006). Perhaps global classification of tumor cells with high-density arrays will become part of a prognostic or predictive molecular signature panel in the future.
Specific Gene Mutations
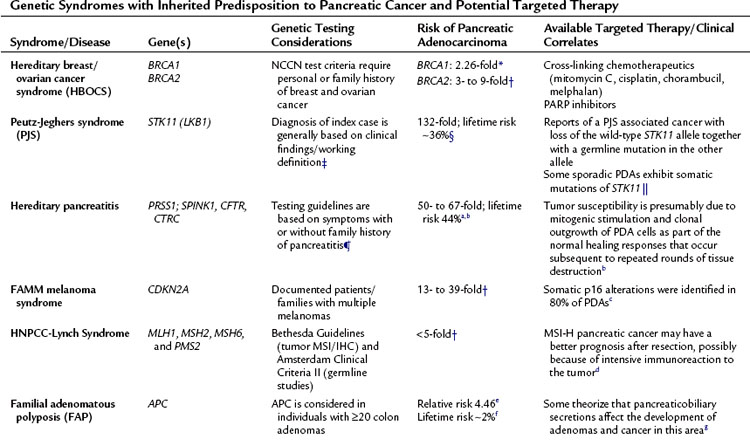
Much like other solid tumors, genes altered in pancreatic cancer include three functional classes: oncogenes, tumor suppressor genes, and caretaker genes. A family of caretaker genes recognized as being disrupted in pancreatic cancer are genes of the Fanconi anemia complementation group, which are involved in homologous recombination-based DNA damage repair (D’Andrea et al, 2003). Patients with Fanconi anemia present with a wide variety of clinical issues, including aplastic anemia and a high risk of developing cancer. BRCA2 is a member of this DNA repair pathway and is mutated in a subset of familial pancreatic cancers (Murphy et al, 2002). This has led to the search for mutations in other Fanconi anemia genes in pancreatic cancer. Somatic mutations of two genes in the core complex, FANCC and FANCG, were discovered but are rare in sporadic pancreatic cancers (van der Heijden et al, 2003). Through other modern techniques, Jones and colleagues (2009) discovered FANCN (PALB2) as another mutated gene found in familial pancreatic cancers.
Mutations in this core complex and in this DNA repair mechanism have major therapeutic implications (Fig. 8A.5, Table 8A.1; see the Familial Pancreatic cancer section below) (van der Heijden et al, 2004). Unlike most in vivo experiments, xenografted mice with isogenic cell lines (FANCC deficient and proficient) experienced regression of tumor after a single dose of the available mitomycin C intrastrand cross-linking drug (van der Heijden et al, 2005). Although other mutations in the Fanconi complementation group have not yet been described—other than FANN, FANCC, and FANCG—and the frequency of these mutations in PDA appear to be low, it is likely that defects in other FANC genes yet to be thoroughly investigated (i.e., FANCA) are the direct cause of some familial and sporadic PDAs.
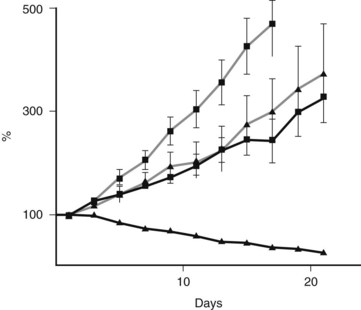
FIGURE 8A.5 Preclinical model shows an example of a successful targeted treatment strategy against a Fanconi-deficient tumor. A single-dose treatment with mitomycin C (5 mg/kg) of pancreatic cancer cell lines xenografted into nude mice. Note the hypersensitivity and tumor regression in the FANCC-deficient PL11 cells (squares) compared with the retrovirally corrected FANC-proficient PL11 cells (triangles). Solid lines indicate treated mice; gray lines indicate no treatment controls. Similar sensitivity was seen in the BRCA2-deficient CAPAN1 xenografted cells (van der Heijden et al, 2005).

Oncogenes
Perhaps the best evidence that KRAS activation is an early and important event in tumorigenesis comes from decades of research on pancreatic cancer. The KRAS2 oncogene on chromosome 12p is the most commonly altered oncogene, with up to 90% of pancreatic cancers containing mutations on codons 12, 13, and 61 (Caldas et al, 1994b). Activating mutations impairs the intrinsic GTPase activity of the KRAS2 gene product, resulting in a protein that is constitutively active in intracellular signal transduction. Inactivation of KRAS2 happens early in the pathway to oncogenesis, with approximately 30% of PanIN-1 lesions harboring KRAS2 mutations (Hruban et al, 2000; Moskaluk et al, 1997). The first mouse model of pancreatic cancer was generated by constitutive overexpression of mutant KRAS2 in the murine pancreatic ductal epithelium, underscoring its importance in pancreatic oncogenesis (Hingorani et al, 2003). This model was further developed into a powerful and useful preclinical model for PDA progression (Hingorani et al, 2005). Several good sources on mouse modeling and pancreatic cancer have been published (Frese et al, 2007; Karreth et al, 2009; Olive et al, 2006; Tuveson et al, 2005).
Rarely, pancreatic cancers with wild-type KRAS2 genes harbor point mutations of BRAF, another gene in the RAS/RAF/MAP kinase signaling pathway, thereby explaining why mutations of these genes occur in mutually exclusive patterns in pancreatic cancer (Calhoun et al, 2003). This highlights the importance of identifying different molecular targets that lead to similar pathways in pancreatic cancer development and of finding a drug that can target one pathway, not one gene. Studies have shown that targeting KRAS may have potential in modulating angiogenesis in tumorigenesis. Matsuo and colleagues showed that oncogenic overexpression of KRAS increases production of angiogenesis, promoting CXC chemokines and VEGF from human pancreatic duct epithelial cells. This upregulation acts through the mitogen-activated protein kinase pathway and c-JUN signaling (Matsuo et al, 2009). KRAS mutation has been shown to be associated with increased VEGFA expression and poorer prognosis in pancreatic carcinoma (Ikeda et al, 2001). Yet to date, targeting KRAS activation in PDA patients has shown no success. Perhaps KRAS activation is a critical early event in pancreatic tumorigenesis, but once cells become malignant, there is no need for constitutive KRAS activation or for oncogenic addiction for that matter.
Other oncogenes implicated in pancreatic cancers include MYC and EGFR, which can be amplified in various subsets of cancers. Overexpression of MYC transcripts occurs in approximately 50% to 60% of pancreatic cancers (Aguirre et al, 2004; Han et al, 2002). Therapeutic targeting using monoclonal antibodies or small-molecule tyrosine kinase inhibitors makes use of the constitutive activation of the EGFR signaling pathway in pancreatic cancers via amplification of the EGFR gene (Holzmann et al, 2004; Li et al, 2004). Lack of a rigorous and reliable immunohistochemical assay to detect overexpression of EGFR has proven to be difficult and limiting.
Tumor Suppressor Genes
CDKN2A, on chromosome 9p, is the most commonly inactivated gene in pancreatic cancers, occurring in 90% of patients (Caldas et al, 1994a; Schutte et al, 1997). CDKN2A belongs to the cyclin-dependent kinase inhibitor family and inhibits cell-cycle progression through the G1-S checkpoint mediated by cyclin-dependent kinases such as CDK4 and CDK6. Homozygous deletions (40%), intragenic mutation with loss of the second allele (40%), and epigenetic silencing by promoter methylation (10% to 15%) all contribute to gene inactivation. Immunolabeling for nuclear p16 protein expression is a reliable surrogate for CDKN2A genetic status; however, such immunolabeling does not identify specific genetic mechanisms of inactivation (Geradts et al, 2000). Loss of CDKN2A function occurs throughout the process of oncogenesis, with lesions appearing in different PanINs: 30% of PanIN-1A and PanIN-1B, 55% of PanIN-2, and 71% of PanIN-3 lesions show loss of nuclear p16 protein expression (Wilentz et al, 1998). The CDKN2A homozygous deletions encompass the MTAP gene in approximately 30% of pancreatic cancers, which offers potential therapeutic benefit, because targeted therapies have been developed that specifically inhibit the growth of MTAP-deficient cells (Chen et al, 1996).
Related posts:
 Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
 Nonhepatic surgery in the cirrhotic patient
Nonhepatic surgery in the cirrhotic patient
 Liver transplantation for cholangiocarcinoma and other neoplastic diseases
Liver transplantation for cholangiocarcinoma and other neoplastic diseases
 Distal splenorenal shunt
Distal splenorenal shunt
 Interventional endoscopy: Technical aspects
Interventional endoscopy: Technical aspects
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


