Fig. 1
Platelet COX-1 mechanism of colon tumorigenesis. Platelets may play an early role in colon tumorigenesis through the release of soluble factors and/or microparticles and exosomes), which can activate stromal cells (macrophages and fibroblasts) translating into an over-expression of COX-2 and the release of prostanoids and growth factors. They may induce intestinal epithelial cell transformation, in part as a consequence of COX-2 expression. Later, in tumor progression there will be the induction of COX-2 also in endothelial cells and this will contribute to a proangiogenic response. Abbreviations: microparticle (MP), endothelial cells (EC), sphingosine-1-phosphate (S1P)
The finding that aspirin benefit was greatest for cancers of the proximal colon (Rothwell et al. 2010) may suggest that platelet-induced tumorigenesis is the mechanism involved in the initiation and/or progression of proximal colon carcinoma. Previous data provide strong support for the hypothesis that proximal and distal colon carcinoma might differ in mechanisms of their initiation and/or progression possibly because the proximal and the distal colon have different embryonic origins and a different vascular supply (Delattre et al. 1989; Iacopetta 2002).
Finally, in this chapter we will discuss COX-independent mechanisms possibly involved in aspirin chemoprevention and we will verify their clinical relevance by considering the available information of pharmacokinetic and pharmacodynamic of aspirin at clinical doses.
2 Mechanisms of Action of Aspirin
2.1 Historical Overview of Aspirin and its Mechanism of Action (Box. 1)
Salicylates, in the form of willow bark, were used as an analgesic during the time of Hippocrates, and their antipyretic effects have been recognized for more than 200 years (Stone 1763). Acetyl salicilyc acid (ASA, aspirin), was introduced in the late 1890s (Dreser 1899) and has been used to treat a variety of inflammatory conditions; however, the antiplatelet activity of this agent was not recognized until almost 70 years later (Weiss and Aledort 1967). Insight into the molecular mechanism of action of aspirin was provided by Gerry Roth and Phil Majerus who used aspirin labeled with 3H at the acetyl group to demonstrate acetylation of prostaglandin (PG) H-synthase (also known as COX) and its irreversible inactivation by the drug (Roth and Majerus 1975; Roth et al. 1975). Importantly, the structural basis of the enzyme inactivation, inferred from the crystal structure of inactivated PGH-synthase, is the blockade of the COX channel in consequence of the acetylation by aspirin of a strategically located serine (Ser) residue (Ser529 in human COX-1) which prevents access of the substrate to the catalytic site of the enzyme (Picot et al. 1994) (Box 1). The discovery of a second isoform of PGH-synthase, called COX-2 (Xie et al. 1991; Kujubu et al. 1991), induced in response to inflammatory and mitogenic stimuli allowed to identify another mechanism of action of aspirin through the acetylation at Ser516 in human COX-2 (Lecomte et al. 1994) (Fig. 2).
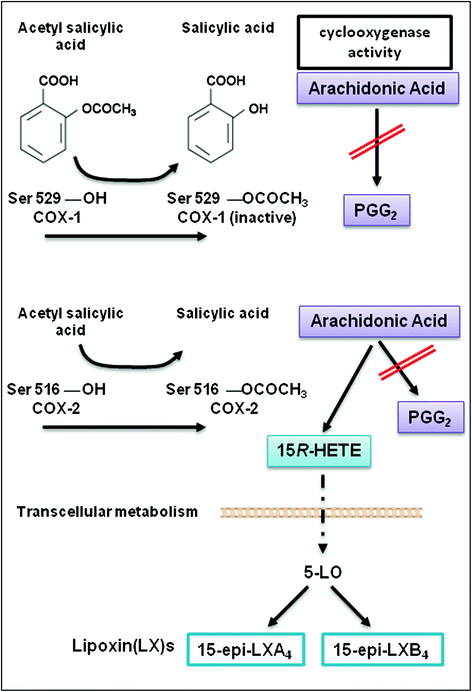
Fig. 2
The molecular mechanism of irreversible inactivation of the cyclooxygenase activity of COX-1 and COX-2 by aspirin through the acetylation of a strategically located serine residue (i.e, Ser529 in the human COX-1 and Ser516 in the human COX-2). In COX-2 expressing cells (such as endothelial cells, leukocytes and epithelial cells), the acetylation of the enzyme by aspirin inhibits its cyclooxygenase activity (preventing the generation of PGG2) but arachidonic acid can be metabolized to 15(R)-hydroxyeicosatetraenoic acid (HETE). 15(R)-HETE can be transformed to 15(R)-epilipoxin(LX)A4 and 15epi-LXB4 in leukocytes via 5-lipoxygenase (5-LO)
Box 1 Historical overview of aspirin and its mechanism of action |
|---|
1890: Felix Hoffman discovered a way to acetylating the hydroxyl group on the benzene ring of salicylic acid to form acetylsalicylic acid (Vane and Botting 2003). |
1971: The work of different scientists demonstrated that aspirin’s mechanism of action is the inhibition of prostaglandin generation: Vane JR (in guinea pig lung cell homogenates) (Vane 1971), Smith JB and Willis AL (in human platelets) (Smith and Willis 1971), Ferreira SH, Moncada S and Vane JR (in spleen) (Ferreira et al. 1971) and Collier JC and Flower RJ (in human seminal vescicles) (Collier and Flower 1971). |
1975: Roth G and Majerus P, using 3H-aspirin, demonstrated acetylation of prostaglandin(PG)-synthase at hydroxyl group of Ser530 and its irreversible inactivation by the drug (Roth and Majerus 1975). |
1976: Hemler M, Lands WEM and Smith WL isolated a ~70 kDa homogeneous, enzymatically active cyclooxygenase (COX) (Hemler et al. 1976). |
1991: Simmons D’s group discovered a distinct COX gene which could be induced with mitogens, growth factors, tumor promoters and lipopolysaccharide, and the induction of which could be inhibited with glucocorticoids. This gene encodes COX-2 protein (Xie et al. 1991); Herschman HR et al. identified the structure of the mitogen-inducible TIS10 gene and demonstrated that the TIS10-encoded protein is a functional prostaglandin G/H synthase (Kujubu et al. 1991). |
1994: Lecomte M & Smith WL demonstrated acetylation of COX-2 at hydroxyl group of Ser 516 and its irreversible inactivation by aspirin (Lecomte et al. 1994); Picot D, Loll P, Garavito M determined the three-dimensional structure of prostaglandin H2 synthase-1 by X-ray crystallography (Picot et al. 1994). |
1996: Kurumbail’s group reported the structures of murine COX-2; these structures explained the structural basis for the selective inhibition of COX-2 (Kurumbail et al. 1996). |
2.2 Insights into the Mechanism of Inhibition of COXs by Aspirin
Aspirin is the only nonsteroidal anti-inflammatory drug (NSAID) which causes an irreversible inactivation of COX-1 and COX-2 by acetylation of a specific serine moiety (Ser529 of COX-1 and Ser516 of COX-2) (Loll et al. 1995; Picot et al. 1994; Lecomte et al. 1994) (Fig. 2).
Both COX-1 and COX-2 catalyze the conversion of arachidonic acid (AA) to prostanoids [prostaglandin (PG)E2, PGF2α, PGD2, prostacyclin(PGI2), and thromboxane(TX)A2] (FitzGerald 2003) (Fig. 3). COX-1 gene is considered a “housekeeping gene” and it is highly expressed in platelets and gastric epithelial cells where it plays a role in causing platelet activation, via the generation of TXA2, and gastric cytoprotection, via the generation mainly of PGE2, respectively (Patrono et al. 2001; Capone et al. 2007; Smyth et al. 2009). Differently, the gene for COX-2 is a primary response gene with many regulatory sites (Kang et al. 2007). However, COX-2 is constitutively expressed in some cells in physiologic conditions, such as endothelial cells (Topper et al. 1996), where COX-2-dependent-PGI2 induces an antithrombtic and vasoprotective signaling (Grosser et al. 2006; Di Francesco et al. 2009), and in pathological conditions, such as cancer cells where the major product is PGE2 (Dixon et al. 2001). COX-2 overexpression, in cancer cells, occurs through post-transcriptional mechanisms, in part due to altered expression of trans-acting factors that bind to AREs (AU-rich) elements and regulate the status of mRNA stability (Harper and Tyson-Capper 2008) (see Chap. 2 for a detailed description by Dixon et al.).
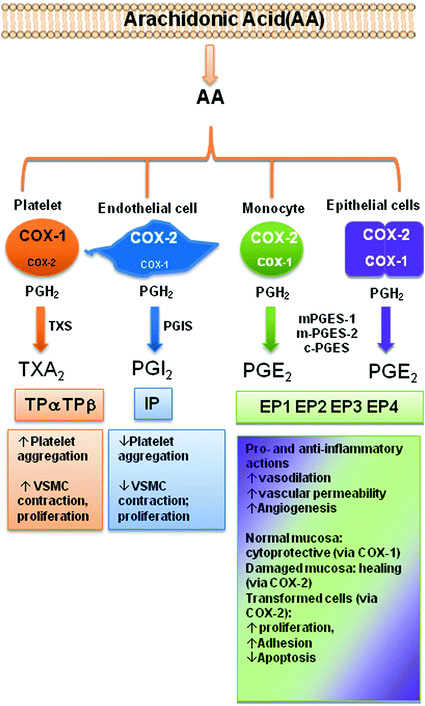
Fig. 3
The product of cyclooxygenase activity of COX-1 and COX-2, PGH2 is metabolized to different prostanoids by tissue-specific synthases. Released prostanoids regulate different functions by the interaction with G-protein coupled receptors. Abbreviations: COX cyclooxygenase; TXA2 synthase (TXS), PGI2 synthase (PGIS), microsomal (m) or cytoplasmatic (c), PGE2 synthase (PGES), TXA2 receptor (TP), PGI2 receptor (IP), EP1-4 (PGE2 receptor)
Both the COX-isozymes have two catalytic activities (Fig. 4): (1) a COX activity responsible for oxygenating AA to PGG2; and (2) a peroxidase (POX) activity that catalyzes a two-electron reduction of PGG2 to PGH2.
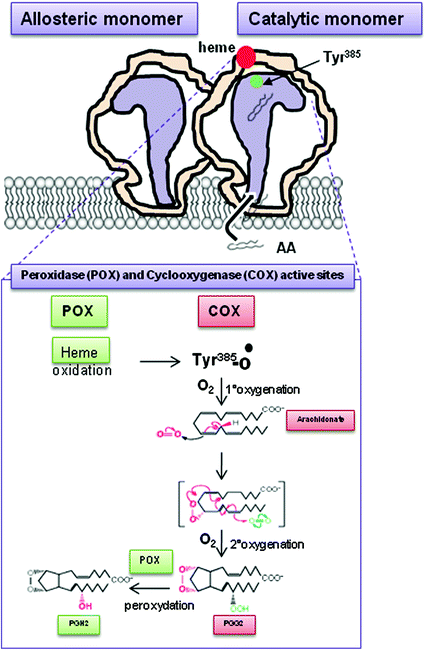
Fig. 4
Mechanism of PGH2 generation by COX-1 and COX-2. Both COX-1 and COX-2 possess peroxydase (POX) and cyclooxygenase (COX) activities. Thus, COX-isozymes generate the same product PGH2. The first step is the oxidation of the heme group at the POX active site, which causes the formation of a tyrosyl radical at Tyr385 in COX active site. The Tyr385 radical stereospecifically abstracts a hydrogen atom from carbon13 of arachidonic acid (AA)(1st oxygenation), then the carbon radical induces the formation of an oxane ring. The addition of a second O2 molecule at carbon 15 ultimately produces PGG2 (2nd oxygenation). PGG2 is the substrate of POX activity which converts peroxide group of PGG2 to hydroxyl forming PGH2
COX-1 and COX-2 are homodimers that exhibit half of sites with COX activity, with AA as the substrate (Yuan et al. 2006; Sidhu et al. 2010) (Fig. 5): only one monomer is able to catalyze a reaction at a given time. The noncatalytic monomer functions as an allosteric regulator of the catalytic monomer (Kulmacz and Lands 1985; Yuan et al. 2006, 2009). This is of potential importance in vivo, where certain fatty acids can function as allosteric regulators of COXs, inhibiting COX-1 and stimulating COX-2 (Yuan et al. 2009).
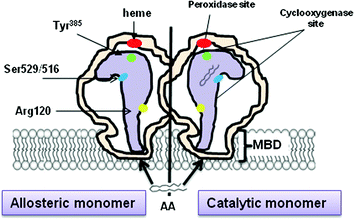
Fig. 5
Dimeric structure of COXs. COX-1 and -2 are homodimers composed of 2 monomers (about 72 kDa) that are tightly bonded to one another. Each monomer is anchored to the cellular membrane through a membrane binding domain (MBD) and contains both cyclooxygenase (COX) and peroxidare (POX) sites
Aspirin binds to one monomer of COX-1 and -2 by the interaction with Arg120 residue and modifies covalently COX isoenzymes by the acetylation of Ser529 and Ser516 on COX-1 and COX-2, respectively; the acetylated monomer becomes the allosteric subunit, and the partner monomer becomes the catalytic monomer. Acetylation of the allosteric subunit of COX-1 causes an irreversible inactivation of the COX activity of the enzyme which translates into the inhibition of the generation of PGG2 from AA (Fig. 2). In other words, aspirin acts as negative allosteric effector by binding to one monomer of COX-1, thus markedly reducing or even eliminating the activity of the partner monomer (Rimon et al. 2010).
Aspirin inhibits COX-2 activity, in a concentration-dependent fashion, through the acetylation of a single monomer of the enzyme (Sharma et al. 2010) (Fig. 2). However, the outcome is somewhat more complex than that seen with COX-1. The acetylated COX-2 has a significantly compromised ability to form PGG2 but produces an alternative product, 15R-hydroxyeicosapentaenoic acid (15R-HETE) from AA (Lecomte et al. 1994). Sophisticated experiments performed by Smith’s group (Sharma et al. 2010) showed that aspirin acetylation of the regulatory monomer of COX-2 is associated with an irreversible inhibition of the catalytic monomer to form PGG2. In contrast, the acetylated monomer forms primarily 15R-HETE from AA (Fig. 2). Thus, the effect of aspirin on COX-2 is an incomplete allosteric inhibitory effect compared with that seen with COX-1 (Sharma et al. 2010).
Several studies in vitro have shown that 15R-HETE is then metabolized to the epi-lipoxins (LXs) in monocytes and leukocytes through the action of 5-lipoxygenase (5-LOX) (Fig. 2) (Gilroy 2005; Serhan 2005), the enzyme also responsible for initiation of leukotriene synthesis. The epi-LXs may cause antiproliferative and anti-inflammatory actions (Serhan 2005; Fierro et al. 2002; Romano 2010). However, convincing evidences that these lipid mediators triggered by aspirin are generated in vivo in humans are lacking. In particular, the analytical assays (mainly immunoassays) (Romano 2006) used to measure their levels in urinary collections were not rigorously validated by comparison with mass-spectrometry analysis.
3 Pharmacology/Pharmacokinetic of Aspirin
3.1 COX-Isozyme Selectivity in Vitro
The inhibitory effect of aspirin on platelet COX-1 activity is evaluated by assessing TXB2 generated during whole blood clotting for 1 h at 37 °C (serum TXB2) (Patrono et al. 1980). As shown in Fig. 6a, aspirin affects platelet COX-1 activity with an IC50 value (concentration which inhibits by 50 % the activity of COXs) of 18 μM. In heparinized human whole blood incubated for 24 h with bacterial endotoxin LPS (which causes a time-dependent induction of COX-2 mainly in monoctyes) (Patrignani et al. 1994), aspirin inhibits PGE2 generation with an IC50 value of approximately 5 mM (Fig. 6a). Under these experimental conditions, salicylic acid (the hydrolysis product of aspirin) affects platelet COX-1 and monocyte COX-2 activities with comparable IC50 values of approximately 1 mM (Fig. 6b). This may suggest the contribution of salicylic acid to the inhibition of COX-2 in whole blood by aspirin, assessed at 24 h of incubation. In fact, aspirin is unstable in blood and it can be deacetylated to salicilic acid by the activity of plasma esterases. We have shown that in human whole blood, aspirin loses its capacity to inhibit platelet COX-1, in a time-dependent fashion, with a t1/2 of approximately 2 h (Fig. 7) (Cipollone et al. 1997). In order to avoid the influence of aspirin metabolism in plasma to its potency to affect COX-isozymes, we assessed aspirin inhibitory effects in washed human platelets (expressing only COX-1) and isolated human monocytes (expressing COX-2 after overnight incubation with LPS), incubated at 37 °C for 60 min with a low concentration of AA, i.e. 0.5 μM. Under these experimental conditions, aspirin results 60-fold more potent to inhibit platelet COX-1 than monocyte COX-2 (Fig. 8).
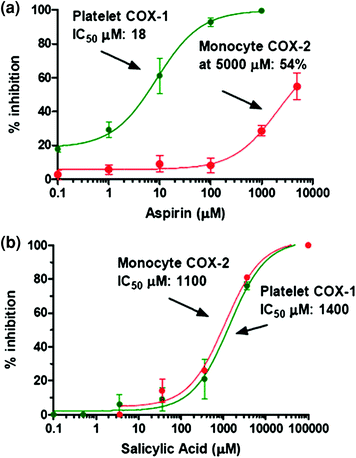
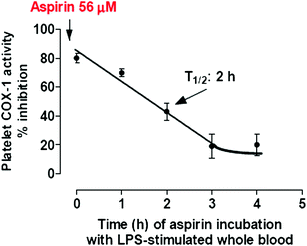
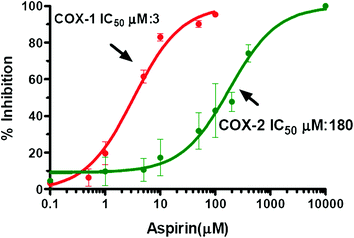
Fig. 6
Inhibitory effects of Aspirin and Salicylic Acid on platelet COX-1- and monocyte COX-2-dependent prostanoid biosynthesis in human whole blood. a Increasing concentrations of Aspirin (0.1–5,000 μM) were used and concentration–response curves for inhibition of platelet COX-1 activity (in green; assessed by measuring TXB2 levels in an aliquot of human whole blood allowed to clot for 1 h at 37 °C) and of monocyte COX-2 activity (in red, assessed by measuring of PGE2 in LPS-stimulated human whole blood for 24 h) were depicted. b Concentration–response curves for inhibition of platelet COX-1 and monocyte COX-2 by increasing concentration of Salicylic acid (0.1–5,000 μM). IC50 values were reported for all curves with the exception of the inhibition curve of monocyte COX-2 by Aspirin in which the maximun inhibition value reached with 5,000 μM of aspirin was 54 %. Concentration–response curves were fitted, and IC50 values were analysed with PRISM (GraphPad, San Diego, CA, USA)
Fig. 7
Time-dependent deacetylation of aspirin in blood. Aspirin was incubated with 1-mL aliquots of heparinized blood samples for 0, 1, 2, 3 and 4 h at 37 °C in the presence of LPS (10 μg/mL). At the end of each incubation, plasma was separated by centrifugation and an aliquot of plasma (corresponding to 56 μM of aspirin) were immediately added to 1-mL samples of whole blood that were allowed to clot at 37 °C for 60 min; serum TXB2 levels were then measured. The half-life of the loss of inhibitory capacity of platelet COX-1 activity was reported
Fig. 8
Effects of Aspirin on prostanoid biosynthesis by washed human platelets (expressing COX-1) and isolated monocytes (expressing COX-2). Concentration–response curves for inhibition of platelet COX-1 and monocyte COX-2 were obtained by the treatment of washed human platelets (expressing only COX-1) and isolated human monocytes (previously incubated overnight with LPS to induce COX-2) with increasing concentrations of Aspirin (0.1–5,000 μM) and AA (0.5 mM) for 60 min at 37 °C. The inhibition of platelet COX-1 activity was assessed by measuring TXB2 levels, while monocyte COX-2 activity was assessed by measuring PGE2 levels. Concentration–response curves were fitted, and IC50 values were analysed with PRISM (GraphPad, San Diego, CA, USA)
3.2 Pharmacokinetic
Aspirin is rapidly absorbed in the stomach and primarily in the upper intestine. Peak plasma levels occur 30–40 min after aspirin ingestion. In contrast, it can take up to 3–4 h to reach peak plasma levels after administration of enteric-coated aspirin (Patrono et al. 2008). The inhibition of platelet COX-1 activity (assessing serum TXB2) ex vivo (after oral dosing) is detectable before aspirin reaches the systemic circulation which is consistent with the inhibition of platelet COX-1 in the presystemic (portal) circulation (Pedersen and FitzGerald 1984). This is further sustained by the fact that the administration of low-dose aspirin (75–100 mg) causes an almost complete suppression of serum TXB2 (>95 %) (Patrignani et al. 1982) which cannot be explained by circulating plasma concentrations of aspirin (approximately 7 μM; Table 1) (Charman et al. 1993). In fact, as shown in Fig. 6a, an almost complete inhibition of platelet COX-1 can be obtained by adding 100 μM of aspirin to whole blood aliquots. The inhibition of platelet function is evident by 1 h after oral dosing with aspirin (Patrono et al. 1985).
Table 1
Peak plasma concentrations (Cmax) of aspirin and salicylic acid after the administration of different doses of aspirin
Aspirin doses | Aspirin Cmax | Salicylic acid Cmax |
|---|---|---|
Antiplatelet dose, 75 mg/day Solution Controlled release | 7.31 μMa 0.29–0.54 μMa | 15 μM 4 μM |
Analgesic doses, 325–600 mg/4–6 h | 28–80 μMb,c | 500 μMd (1 g single dose) |
Anti-inflammatory dose, 1.2 g/4–6 h | 142 μMc | 1500–2500 μMd,e |
The plasma half-life of aspirin is only 20 min; however, because platelets have a limited capacity to generate COX-1 de novo (Evangelista et al. 2006), the irreversible inhibition on platelet COX-1 by aspirin lasts for the duration of the life of the platelet (i.e. 10 days) (Patrignani et al. 1982). For all these reasons, the oral administration of aspirin at low-doses, once daily, causes an almost complete suppression of platelet COX-1 which persists throughout dosing interval (Patrignani et al. 1982). This is a fundamental requisite to obtain an antithrombotic effect (Patrono et al. 2008). In fact, even tiny concentrations of TXA2 can activate platelets and they can synergize with low-concentrations of other agonists to cause a complete platelet aggregation (Minuz et al. 2006).
The oral bioavailability of regular aspirin tablets is approximately 40–50 % over a wide range of doses (Pedersen and FitzGerald 1984). A considerably lower bioavailability has been reported for enteric-coated tablets and sustained-release, microencapsulated preparations (Charman et al. 1993; Clarke et al. 1991). Because platelet COX-1 is acetylated in the presystemic circulation, the antiplatelet effect of aspirin is largely independent of systemic bioavailability (Pedersen and FitzGerald 1984).
3.3 Determinants of Achieved COX-Isozyme Selectivity by Clinical Doses of Aspirin
The administration of low-dose aspirin is associated with a preferential inhibition of platelet COX-1 ex vivo (Patrignani et al. 1982; Patrono et al. 1985). In fact, the levels of aspirin (approximately 7 μM) detectable in the systemic circulation (Charman et al. 1993) can cause only a trivial (Figs. 6a and 8) and reversible inhibition of COX-2 expressed in nucleated cells (for de novo synthesis of the acetylated protein). In contrast, platelet COX-1 is completely and irreversibly inhibited in the presystemic circulation and this effect persists for the interval between doses because platelets are anucleated cell fragments. This platelet COX-1 selectivity can be enhanced by the slow administration of low-dose aspirin. In fact, a controlled-release formulation of aspirin 75 mg with negligible systemic bioavailability (Charman et al. 1993; Table 1) has been shown to achieve selective inhibition of platelet TXA2 production without suppressing systemic PGI2 synthesis (Clarke et al. 1991), which is mainly derived from vascular COX-2 (Grosser et al. 2006), and presumably other prostanoids generated in other nucleated cell types.
Aspirin administered at analgesic (325–600 mg every 4–6 h) and anti-inflammatory (1.2 g every 4–6 h) doses is associated with circulating concentrations in the range of 30–150 μM (Pedersen and FitzGerald 1984; Seymour and Rawlins 1982; Table 1) that may affect COX-2 activity in a dose-dependent fashion (Figs. 6a and 8). Circulating concentrations of aspirin hydrolysis product, i.e. salicylic acid, might contribute to affect COX-2 activity or expression when aspirin is administered at very high doses (>1000 mg) (Smyth and Dawkins 1971; Rumble et al. 1980; Table 1).
3.3.1 Dose-Dependence of Gastrointestinal Toxicity
Observational studies (García Rodríguez et al. 2001, 2011) and a meta-analysis of randomized clinical trials in high-risk patients (Baigent and Collaboration 2002) have demonstrated that long-term therapy with low-dose aspirin approximately doubles the risk of major extracranial (mostly, upper gastrointestinal) bleeding (Patrono et al. 2005). It is likely that these complications are related to inhibition of platelet COX-1 (Patrono et al. 2001). However, the risk of upper gastrointestinal bleeding increases at higher doses of aspirin (García Rodríguez et al. 2001) and this is plausibly due to the contribution of the inhibition of gastrointestinal COX-1 (Fig. 3) by systemic plasma concentrations of aspirin.
4 Role of Platelets in Tumorigenesis
4.1 Platelet-Mediated Mechanisms in Tumorigenesis and Metastasis
Platelets represent an important linkage between tissue damage/dysfunction and the inflammatory response initially acting to repair the damage but, if platelet activation is uncontrolled, this translates into a wide spectrum of pathological conditions, such as atherothrombosis and cancer. Moreover, the role of platelets has been recognized from a long time in the process of spreading of neoplastic cells to other organs or to lymph nodes far from the primary tumor (called metastatic disease) (Gay and Felding-Habermann 2011). Interestingly, Folkman and associates showed that platelets sequester angiogenesis regulatory proteins and that the analysis of the “platelet angiogenesis proteome” could be used for ultra-early detection of recurrent cancer years before it is symptomatic or can be anatomically located (Cervi et al. 2008).
Platelets store and release, after activation, various angiogenic-regulating factors, such as vascular endothelial growth factor (VEGF), PDGF, fibroblast growth factor (FGF), insulin-like growth factor (IGF), endostatin, thrombospondin-1, tissue inhibitor of metalloproteinases (TIMP). These products are present in different sets of α-granules suggesting a differential release of pro- and anti-angiogenic factors in different districts (Italiano et al. 2008). In addition, platelets can synthesize and release cytokines, such as IL-1β (Dixon et al. 2006). Platelet-derived IL-1β, generated by the interaction of platelets with monocytes, has been shown to contribute to monocytic COX-2 induction through a post-transcriptional mechanism which stabilizes COX-2 mRNA (Dixon et al. 2006).
Activated platelets may play a role in tumor progression and metastasis also by the release of microparticles (MPs) and exosomes (Janowska-Wieczorek et al. 2005). These platelet-derived MPs are approximately 0.1–1.0 μm in diameter in humans and express P-selectin (CD62P) and GP IIb-IIIa. They adhere to a variety of cells, can activate endothelial cells, leukocytes and other platelets, and deliver signals through chemokines (Mause and Weber 2010). Exosomes, range in size from 0.04 to 0.1 μm, arise from the internal membrane vesicles of multivesicular bodies and platelets α-granules. Unlike MPs, exosomes do not share a similar surface phenotype of activated platelets. However, both MPs and exosomes are known to carry and deliver cellular signals, suggesting a potential role in platelet-derived signaling.
Another mechanism by which platelets influence tumorigenesis is through the generation of TXA2, a major product of platelet COX-1, which promotes platelet aggregation and vasoconstriction (Grosser et al. 2006). TXA2 has been reported to be involved in angiogenesis and development of tumor metastasis (Honn 1983). Interestingly, it has been shown that enhanced TXA2 generation by the introduction of the downstream TXA2 synthase (TXAS) into murine colon-26 adenocarcinoma cell line (C26) enhanced tumor growth in vivo through the stimulation of tumor angiogenesis (Pradono et al. 2002). Similarly, TXA2 promotes the interaction between metastasizing tumor cells and the host hemostatic system (Pradono et al. 2002). Thus, pharmacological inhibition of TXA2 synthase (TXAS) has been shown significantly to inhibit tumor cell growth, invasion, metastasis and angiogenesis in a range of experimental models (Honn 1983). However, the recent finding that aspirin reduces the incidence and mortality of CRC, at doses of at least 75 mg daily (Rothwell et al. 2010), recommended for the prevention against heart disease (Patrono et al. 2005), strongly suggests a role of TXA2, derived mainly from platelets, in tumorigenesis (Patrono et al. 2001) (Fig. 1).
Recent findings have shown that platelets generate and store high amounts of sphingosine-1-phosphate (S1P) which is released upon stimulation with activators of protein kinase C (PKC), such as thrombin, but also with a TXA2 mimetic (Ulrych et al. 2011). Once released, S1P may regulate processes such as inflammation, neovascularization, cell growth and survival (Pyne and Pyne 2010). Interestingly, it was found that oral ASA (500 mg single dose or 100 mg over 3 days) attenuated S1P release from platelets in healthy volunteers ex vivo and it was proposed that the inhibition of TXA2 by aspirin might play a role in the depression of S1P release (Ulrych et al. 2011). Aspirin added in vitro to washed human platelets caused a concentration-dependent reduction of S1P release which however, it was not complete even at 300 μM (Ulrych et al. 2011). Thus, aspirin is more potent to inhibit TXB2 than S1P, both ex vivo and in vitro. Further studies should be performed to address whether the partial depression of platelet S1P release ex vivo might contribute to aspirin chemoprevention of CRC.
Related posts:
 Emerging Role for Anti-inflammatory Agents for Chemoprevention
Agents with Anti-lnflammatory Properties in Chemoprevention of Colorectal Neoplasia
Emerging Role for Anti-inflammatory Agents for Chemoprevention
Agents with Anti-lnflammatory Properties in Chemoprevention of Colorectal Neoplasia
 Aspects of COX-2 Expression in Colorectal Neoplasia
Aspects of COX-2 Expression in Colorectal Neoplasia
 NSAID Targets and Derivatives for Colorectal Cancer Chemoprevention
NSAID Targets and Derivatives for Colorectal Cancer Chemoprevention
 Pharmacology, Toxicity and Efficacy in Cancer Clinical Trials
Pharmacology, Toxicity and Efficacy in Cancer Clinical Trials
 Inheritance and Strategies for Prevention in Populations at High Risk of Colorectal Cancer (CRC)
Inheritance and Strategies for Prevention in Populations at High Risk of Colorectal Cancer (CRC)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


