Phase
Definition
0
Exploratory study involving very limited human exposure to the drug, with no therapeutic or diagnostic goals (e.g., screening studies, microdose studies)
1
Studies that are usually conducted with healthy volunteers and that emphasize safety. The goal is to find out what the drug’s most frequent and serious adverse events are and, often, how the drug is metabolized and excreted
2
Studies that gather preliminary data on effectiveness (whether the drug works in people who have a certain disease or condition). For example, participants receiving the drug may be compared with similar participants receiving a different treatment, usually an inactive substance (called a placebo) or a different drug. Safety continues to be evaluated, and short-term adverse events are studied
3
Studies that gather more information about safety and effectiveness by studying different populations and different dosages and by using the drug in combination with other drugs
4
Studies occurring after FDA has approved a drug for marketing. These including postmarket requirement and commitment studies that are required of or agreed to by the sponsor. These studies gather additional information about a drug’s safety, efficacy, or optimal use
Table 17.2
Novel therapies for the treatment of ulcerative colitis
Drug | Mechanism of action | Route | Status |
|---|---|---|---|
Golimumab (Simponi) | TNFα antagonist | SC | Approved for UC |
Biosimilar | TNFα antagonist | ||
Tofacitinib (Xeljanz) | Janus kinase inhibitor | Oral | Phase II completed |
Basiliximab (Simulect) | IL-2 antagonist suppressing lymphocyte activity and reducing corticosteroid resistance | IV | |
Vedolizumab | Specifically targets the α4β7 integrin (inhibits leukocyte adhesion) | IV | Phase III |
PF-547659 | Monoclonal antibody to MAdCAM-1 (inhibits leukocyte adhesion) | IV/SC | Phase I and II ongoing |
AJM300 | α4-integrin inhibitor (inhibits leukocyte adhesion) | Oral | Phase I |
Etrolizumab | β7-integrin inhibitor (inhibits leukocyte adhesion) | IV/SC | Phase II |
Alicaforsen | Decrease in the production of ICAM-1 (inhibits leukocyte adhesion) | Enema | |
BMS-936557 | Antibody to CXCL 10 (chemokine antagonist) | IV | Phase III |
SB-656933 | CXCR2 antagonist (chemokine antagonist) | Phase II terminated; no further drug development | |
Abatacept | Competes with CD28 for CD80 and CD86 binding | IV | |
FMT | Alteration of microbiome | NG, NJ, PR | Clinical trials ongoing |
HMPL-004 | Inhibitory activity against TNF-α, IL-1β, and NF-κB | Oral | Phase II completed |
Treatments Aimed at Blocking Proinflammatory Cytokines
Anti-tumor Necrosis Factor (Anti-TNF) Agents
Golimumab (Formerly Known as CNTO 148)
There are currently several therapies on the market for UC targeting TNF-α including infliximab, adalimumab, and golimumab. Golimumab is a fully humanized monoclonal immunoglobulin also directed against TNF-α. Genetically engineered mice were immunized with human anti-TNFα resulting in an antibody with a human-derived variable and regions that are constant. The variable region of golimumab binds to both the soluble and transmembrane bioactive forms of TNF-α and as a result inhibits the biological activity of TNF-α. Golimumab has been shown in vitro to modulate the biological effects mediated by TNF including the expression of adhesion proteins responsible for leukocyte infiltration (E-selectin, ICAM-1, and VCAM-1) and the secretion of proinflammatory cytokines (IL-6, IL-8, G-CSF, and GM-CSF).
Golimumab has been approved by the Food and Drug Administration in the United States to treat moderately to severely active rheumatoid arthritis (RA), active psoriatic arthritis, and active ankylosing spondylitis (AS) and recently gained regulatory approval in 2013 for the treatment of moderate to severe UC patients who have had an inadequate response or intolerance to prior conventional treatments or who require continuous steroid therapy. Golimumab is given subcutaneously, and for UC the dosage recommended is 200 mg initially at week 0 and then 100 mg at week 2 and then 100 mg every 4 weeks. Serum golimumab concentrations reach steady-state pharmacokinetics by week 8 after the first maintenance dose. Treatment with 100 mg golimumab subcutaneously every 4 weeks during maintenance resulted in a mean steady-state trough serum concentration of 1.8 ± 1.1 μg/ml [1].
A combined double-blind, placebo-controlled, phase II dose-finding and phase III dose-confirmation trials demonstrated golimumab’s efficacy for induction of a clinical response and remission in patients with moderate to severe ulcerative colitis (PURSUIT). There were 1,064 adults with UC (Mayo score, 6–12, endoscopy subscore ≥2). Patients were randomly assigned to groups given golimumab doses of 100 mg and then 50 mg (phase II only), 200 mg and then 100 mg, and 400 mg and then 200 mg, 2 weeks apart. The phase III primary endpoint was a clinical response at week 6. The secondary endpoints included clinical remission, mucosal healing, and IBDQ score change at week 6. In phase II, median changes from baseline in the Mayo score were −1.0, −3.0, −2.0, and −3.0 in placebo and 100 mg/50 mg, 200 mg/100 mg, 400 mg/200 mg golimumab, respectively. In phase III, rates of clinical response at week 6 were 51.8 % and 55 % among patients given 200 mg/100 mg and 400 mg/200 mg golimumab respectively vs. 29.7 % in the placebo group (P < 0.0001). Rates of clinical remission and mucosal healing and mean changes in the IBDQ scores were significantly greater in both the golimumab groups compared to placebo group (P ≤ 0.0005). Rates of serious adverse events were 6.1 % and 3.0 %, and rates of serious infection were 1.8 % and 0.5 % in the placebo and golimumab groups, respectively. One patient in the 400/200 mg group died from surgical complications of an ischiorectal abscess.
In the phase III, double-blind trial evaluating golimumab in the maintenance of a clinical response in patients with moderate to severe UC, patients who responded to the initial golimumab induction therapy were randomly assigned to groups given placebo or injections of 50 or 100 mg of golimumab every 4 weeks through week 52 [2]. Four hundred sixty-four patients were included in this study. Patients who responded to placebo in the induction study continued to receive placebo. Nonresponders in the induction study received 100 mg golimumab. The primary outcome was clinical response maintained through week 54, and secondary outcomes included clinical remission and mucosal healing at week 30 and week 54. Clinical response was found to be maintained in 47.0 % receiving 50 mg golimumab, 49.7%receiving 100 mg golimumab, and 31.2 % receiving placebo (P = 0.010 and P < 0.001, respectively). At weeks 30 and 54, 27.8 % patients who received 100 mg golimumab were in clinical remission, and 42.4 % had mucosal healing compared to placebo (15.6 % and 26.6 %, P = 0.004 and P = 0.002, respectively) or 50 mg golimumab (23.2 and 41.7 %). Serious adverse events occurred in 7.7 %, 8.4 %, and 14.3 % of patients given placebo, 50 mg or 100 mg golimumab, respectively. Serious infections occurred in 1.9 %, 3.2 %, and 3.2 % in placebo, 50 mg or 100 mg golimumab, respectively. Among all patients who received golimumab, three died and four developed active tuberculosis. Of those that died, they all received 100 mg golimumab and died from sepsis, tuberculosis, and cardiac failure.
In controlled phase III trials through week 16 in patients with rheumatoid arthritis, psoriasis, and ankylosing spondylitis, serious infections were observed in 1.4 % of golimumab-treated patients and 1.3 % of control patients. In these trials, the incidence of serious infections per 100 patient years of follow-up was 5.7 (95 % CI: 3.8, 8.2) for the golimumab group and 4.2 (95 % CI 1.8, 8.2) for the placebo group. In the controlled phase II/III trial through week 6 of golimumab induction in UC, the incidence of serious infections in the golimumab 200/100 mg-treated patients was similar to the incidence of serious infections in placebo-treated patients. Through week 60, the incidence of serious infections was similar between patients who received golimumab induction and 100 mg maintenance compared to those who received golimumab induction and placebo for maintenance. Serious infections in golimumab patients included sepsis, pneumonia, cellulitis, abscess, tuberculosis, invasive fungal infections, and hepatitis B infection [1].
During controlled portions of the phase II trial in RA and the phase III trials in rheumatoid arthritis, psoriasis, and ankylosing spondylitis, the incidence of malignancies other than lymphoma per 1,000 patient years of follow-up was not higher in the combined golimumab group compared to placebo group and was similar to that expected in the general US population according to the SEER database. In the phase II/III trials in UC, the incidence of non-lymphoma malignancies was also similar between the drug and placebo groups [1].
In UC trials, 3 % [34], 28 % (341), and 69 % (823) of golimumab-treated patients were positive, negative, and inconclusive for antibody development. No definitive conclusions regarding the relationship between antibodies to golimumab and clinical efficacy or safety measures could be drawn due to small sample size [1].
Anti-TNF Biosimilars
The Biologics Price Competition and Innovation Act (BPCI Act) was signed into law by President Obama as part of healthcare reform (Affordable Care Act) to encourage the development of biosimilars and interchangeable biological products which ultimately may lead to better patient access and a lower cost to consumers [8].
A biosimilar is a biotherapeutic product which is similar in terms of quality, safety, and efficacy to an already licensed reference biotherapeutic product [9]. To establish the biosimilarity, the US FDA requires that clinical studies must show that there are no clinically meaningful differences between the biosimilar and the reference product in terms of safety, purity, and potency. The drugs must be shown to have the same pharmacokinetics and pharmacodynamics and for the most part equivalent efficacy and safety to the reference product.
The BPCI Act provides an abbreviated licensure pathway for biosimilar and interchangeable biological products under section 351(k) of the Public Health Service Act (PHS Act). As a result, a biosimilar that is demonstrated to be highly similar to the reference product may rely on existing scientific knowledge about the safety, purity, and potency of the reference product and as a result may not be required to provide full product-specific nonclinical and clinical data in order to be licensed [8].
The patent of infliximab (the first anti-TNF-α used in UC) expires between 2013 and 2015 which has opened up the market for the development of biosimilar drugs. Thus far, no single biosimilar has been reported to be tested for UC.
Janus Kinase Inhibitors
Tofacitinib (CP 690550)
Janus kinases (JAK) 1, 2, and 3 are extremely important in cytokine signaling that is involved in lymphocyte survival, proliferation, differentiation, and apoptosis [10]. JAK3 is found only in hematopoietic cells and is part of the signaling pathway activated by IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 which is crucial in the activation, function, and proliferation of lymphocytes (Fig. 17.1) [11].
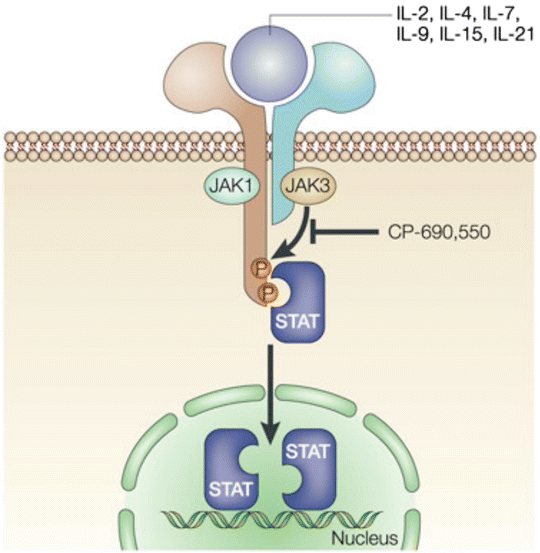
Fig. 17.1
Tofacitinib. With permission from O’Shea, J, Pesu M, Borie DC, Changelian PS. A new modality for immunosuppression: targeting the JAK/STAT pathway. Nature Reviews 2004;4:555–564. © Nature Publishing Group 2004
Tofacitinib is an oral small molecule inhibitor of JAK1 and 3. In vitro studies have shown that it interferes with Th2 and Th17 cell differentiation and blocks the production of IL-17 and IL-22 [12].
In a phase II double-blind, placebo-controlled trial, the efficacy of tofacitinib in 194 adults with moderately to severely active ulcerative colitis was evaluated [13]. Patients were randomly assigned to receive tofacitinib at a dose of 0.5, 3, 10, or 15 mg or placebo twice daily for 8 weeks. The primary outcome was a clinical response at 8 weeks and occurred in 32 %, 48 %, 61 %, and 78 % of patients receiving tofacitinib at a dose of 0.5 mg (P = 0.39), 3 mg (P = 0.55), 10 mg (P = 0.10), and 15 mg (P < 0.001), respectively—compared with 42 % of patients receiving placebo. Clinical remission (Mayo score ≤2 with no subscore >1) at 8 weeks occurred in 13 %, 33 %, 48 %, and 41 % of patients receiving tofacitinib at a dose of 0.5 mg (P = 0.76), 3 mg (P = 0.01), 10 mg (P < 0.001), and 15 mg (P < 0.001), respectively, as compared with 10 % of patients receiving placebo. Treatment with the drug resulted in reduced C-reactive protein and fecal calprotectin levels.
Tofacitinib has generally been well tolerated in clinical trials. The most commonly reported adverse events related to infection reported by Sandborn and colleagues were influenza and nasopharyngitis [13]. Two patients receiving the 10 mg dose twice daily had serious adverse events from infection (postoperative abscess, anal abscess). Of significance but uncertain long-term consequence, a dose-dependent increase in low-density and high-density lipoprotein cholesterol was seen after 8 weeks of treatment which were reversible after discontinuing the studied drugs [13]. Three patients treated with tofacitinib (one at dose of 10 mg twice daily and two at dose of 15 mg twice daily) had an absolute neutrophil count of less than 1,500 (with none being <1,000) [13]. Tofacitinib is a true immunosuppressant and will most likely be used as monotherapy, and there is a concern for increased risk of infections and lymphoma with using this drug compared to other biologics.
There is currently an ongoing multicenter, randomized, double-blind placebo-controlled parallel group study evaluating tofacitinib as a maintenance therapy in patients with ulcerative colitis [14]. Patients will either be given placebo orally twice daily, tofacitinib 5 mg orally twice daily, or tofacitinib 10 mg orally twice daily. The primary endpoint is the proportion of subjects in remission at week 52. Secondary outcomes that will be measured are the proportion of patients with mucosal healing at week 52 and number of patients in sustained steroid free remission [14].
Adjunct Therapy in Corticosteroid Resistance
Basiliximab
Interleukin (IL)-2 is a T-cell autocrine growth factor that has been demonstrated to antagonize the action of steroids, thus contributing to the resistance of lymphocytes to corticosteroids. The high-affinity receptor for IL-2 is CD25 [15–18].
Basiliximab is a chimeric IgG1 monoclonal antibody that binds to the IL-2 receptor (CD25) and has been studied as an agent for the treatment of steroid-resistant ulcerative colitis.
An initial open-label, uncontrolled, 24-week trial was performed with basiliximab in ten patients with steroid-resistant UC. Patients were given a single bolus of 40 mg of intravenous basiliximab plus steroid treatment. The outcomes were assessed using the UC Symptom Score (UCSS), rectal biopsy, and IBDQ. Lymphocyte steroid sensitivity was measured in vitro in 39 subjects in the presence or absence of basiliximab. Nine achieved clinical remission within 8 weeks. Eight of the nine initial responders relapsed (median, 9 weeks), but remission was re-achieved with corticosteroids and azathioprine. Seven patients were in clinical remission at 24 weeks, and five were off of all steroid therapy. In vitro measurement of lymphocyte steroid sensitivity showed steroid resistance in 22 %. All however were rendered steroid sensitive with basiliximab [19].
A further open-label uncontrolled clinical trial was performed in 20 patients with moderate (n = 13) to severe (n = 7) steroid-resistant UC. Patients were given a single dose of 40 mg basiliximab with standard steroid therapy. The primary endpoint was clinical remission within 8 weeks (UCSS). Fifty percent achieved clinical remission within 8 weeks (seven of the moderate, three of the severe) and 65 % at 24 weeks. Five patients required colectomy (four severe and one moderate UC), and one required rescue cyclosporine (moderate UC). Two patients developed herpes zoster. Otherwise, the treatment was well tolerated [20].
However, in contrast, in a study by Sands et al., 149 patients with moderate to severe UC despite treatment with oral prednisone for 14 days were randomly assigned to groups that were given 20 mg (n = 46) or 40 mg (n = 52) basiliximab or placebo (n = 51) at weeks 0, 2, and 4 [21]. All subjects received 30 mg/day prednisone through week 2; the dose was reduced by 5 mg each week to 20 mg/day, which was maintained until week 8. At week 8, rates of clinical remission were compared for patients given basiliximab and placebo. Twenty-eight percent of patients given placebo, 29 % of those given the 40 mg dose of basiliximab, and 26 % of those given the 20 mg dose of basiliximab achieved clinical remission (P = 1.00 vs. placebo for each dose). Six subjects who received basiliximab had serious adverse events (6.1 %) compared with two who received placebo (3.9 %; P = 0.72). Therefore, contrary to the prior open-label trials, this study did not demonstrate the efficacy of basiliximab in increasing the effect of corticosteroids to help induce remission in outpatients with corticosteroid-resistant moderate to severe UC.
Treatments That Target Leukocyte Migration and Adhesion
Adhesion Molecule Blockers
Intercellular adhesion molecules play a role in leukocyte adhesion and migration, local lymphocyte stimulation, and T lymphocyte trafficking in the intestine. These molecules are upregulated in the presence of inflammation.
Vedolizumab (Previously Known as LDP02, MLN02, and MLN0002)
In UC, there is an ongoing inflammatory response with activation of T cells, cytokine production, upregulation of the normal lymphocyte homing response, and recruitment of high numbers of T cells to the intestinal mucosa. The α4β7 integrin molecule is found on circulating T lymphocytes and is involved in the recruitment of leukocytes to the gastrointestinal tract. The α4β7 integrin is activated on the lymphocyte surface membrane and binds with its ligand (mucosal addressin cell adhesion molecule-1—MAdCAM-1) on the endothelial cell surface membranes. This ligand binds lymphocytes from the endothelial lumen as they pass, and once bound, the lymphocytes migrate into the lamina propria and tissue. Studies have shown significantly higher levels of α4β7 integrin and MAdCAM-1 in colons of IBD patients. The binding of these molecules results in the homing of gut-associated lymphocytes to areas of inflamed and normal colonic mucosa (Figs. 17.2 and 17.3).
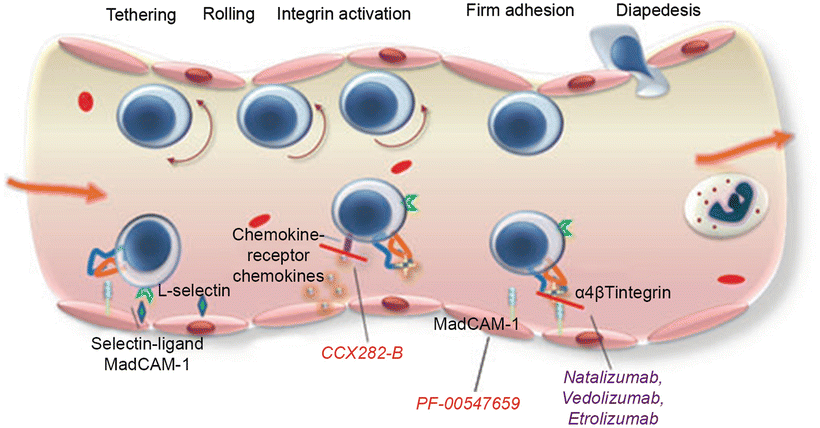
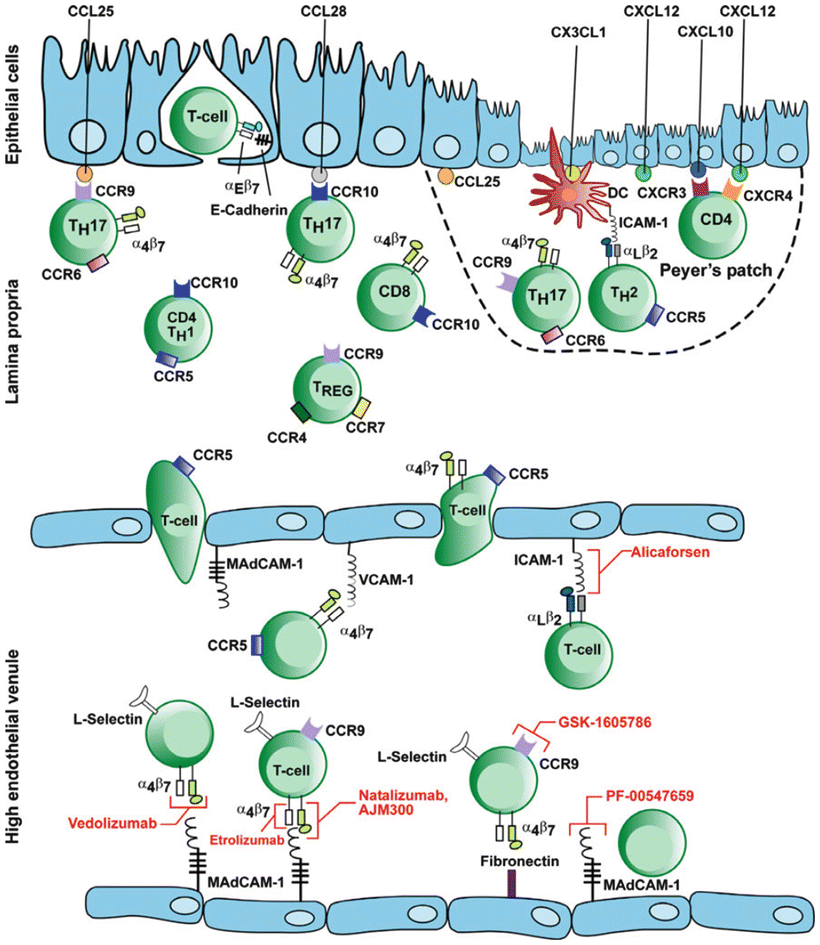
Fig. 17.2
Mechanism of action of adhesion molecules in the intestinal endothelium and their blockage by anti-adhesion drugs. With permission from Lobatón T1, Ve`rmeire S, Van Assche G, Rutgeerts P. Review article: anti-adhesion therapies for inflammatory bowel disease. Aliment Pharmacol Ther. 2014 Mar;39(6):579–94. © 2014 John Wiley & Sons Ltd. [35]
Fig. 17.3
Mechanism of action of adhesion molecules in the intestinal endothelium and their blockage by anti-adhesion drugs. With permission from Thomas S, Baumgart DC. Targeting leukocyte migration and adhesion in Crohn’s disease and ulcerative colitis. Inflammopharmacol 2012;20:1–18. © Springer 2012 [33]
Natalizumab is a humanized monoclonal antibody directed against the α4 integrin on leukocytes (specifically lymphocytes). Because it is a nonselective inhibitor of the alpha 4 integrin which directs leukocytes not only to the intestinal mucosa but also to the central nervous system, there has been a fatal rarely occurring side effect of the drug—the risk of progressive multifocal leukoencephalopathy.
By contrast, vedolizumab is a recombinant IgG1-humanized monoclonal antibody that specifically targets the α4β7 integrin heterodimer which is expressed in essence exclusively in the gut. This selectively blocks the interaction between α4β7 and MAdCAM-1 in the gut, thereby inhibiting leukocyte migration to the intestinal mucosa. Vedolizumab is administered intravenously every 4 weeks.
In 1996, a monoclonal antibody against α4β7 integrin was used to demonstrate resolution of colitis in cotton-top tamarin monkeys. A monoclonal antibody to the α4β7 integrin or a nontherapeutic monoclonal antibody via intramuscular injection was given to eight monkeys with chronic colitis. The control animals showed no improvement, but those receiving the antibody demonstrated clinical and histological response. Animal studies have also shown that inhibition of MAdCAM-1 prevents the development of ileitis in mice by preventing T-cell adhesion to ileal endothelium [22].
In 2000, a phase I double-blind, placebo-controlled trial using humanized α4β7 antibody was reported in abstract form [23]. There were 29 patients with moderately severe UC. A single dose of the humanized antibody was given to participants consisting of 0.15 mg/kg subcutaneously (SC), 0.15 mg/kg intravenously (IV), 0.5 mg/kg IV, and 3 mg/kg IV or placebo. A dose of 0.5 mg/kg was found sufficient to completely saturate the antibody receptors. Complete endoscopic and clinical remission were seen 40 % (two patients) receiving 0.5 mg/kg.
A phase II randomized, double-blind, placebo-controlled trial using the α4β7 antibody (MLN02) was performed over 6 weeks evaluating the drug’s efficacy [24]. One hundred eighty-one adults with moderately or severely active UC (disease extent >25 cm from anal verge, UC clinical score (UCCS) 5–9 points with a score of at least 1 for stool frequency or rectal bleeding, and a modified Baron score of at least grade 2 on sigmoidoscopy) were randomly assigned to receive either placebo, MLN02 0.5 mg/kg IV, or MLN02 2 mg/kg IV. Each patient received two infusions (day 1 and day 29). At week 6, the primary outcome of clinical remission (UCCS of 0–1 and a modified Baron grade of 0–1 with no evidence of rectal bleeding) was seen in 14 %, 33 %, and 32 % in the placebo, 0.5 mg/kg (P = 0.02), and 2 mg/kg (P = 0.02) groups, respectively. Secondary outcomes of clinical response were also higher in the MLN02 0.5 mg/kg and 2 mg/kg groups compared to the placebo groups with 66 %, 53 %, and 33 %, P = 0.002, respectively. Endoscopic remission was seen in 28 %, 12 %, and 8 % of those receiving 0.5 mg/kg, 2 mg/kg, and placebo, respectively.
Later, a phase II randomized controlled trial evaluating increasing doses of vedolizumab was performed using a new formulation developed from using the Chinese hamster ovary cell-based system [25]. Patients were randomized to receive 2, 6, or 10 mg/kg of the new drug or placebo. Patients received four infusions—one on days 1, 15, 29, and 85. Safety, pharmacokinetics, pharmacodynamics, and immunogenicity evaluations were performed at multiple time points to day 253. Forty-six patients (9 placebo and 37 vedolizumab) received at least one dose of study medication. Vedolizumab maximally saturated the a4b7 receptors on peripheral serum lymphocytes at all measurable serum concentrations. Vedolizumab was well tolerated. At every assessment from day 29 to 253, >50 % of the vedolizumab-treated patients were in clinical response; placebo response rates were 22–33 %. After this study was completed, some patients were then assigned to receive vedolizumab 2 or 6 mg/kg every 8 weeks for an additional 547 days, and the efficacy of the drug was assessed using the partial Mayo score (PMS), and the safety, immunogenicity, and pharmacokinetics were analyzed [26, 27]. Eighty-one percent of patients continued on vedolizumab until day 547. Five patients withdrew due to lack of efficacy; three patients withdrew due to adverse events. Remission rates were between 70 and 80 %.
The phase III randomized, double-blind, placebo-controlled trial assessing the efficacy and safety of vedolizumab in induction therapy in patients with moderate to severe UC (Mayo score of ≥6 and an endoscopic subscore of ≥2 despite steroids, thiopurines, and/or anti-TNF therapy) was then performed (GEMINI I) [28, 29]. Patients were randomized to receive either vedolizumab 300 mg IV or placebo on days 1 and 15. Two hundred twenty-four patients received vedolizumab and 149 patients received placebo. Of note, 39 % in the vedolizumab group had previous anti-TNF failure. The clinical response, remission, and mucosal healing rates were 25.5 % vs. 47.1 % (P < 0.0001), 5.4 % vs. 16.9 % (P = 0.0009), and 24.8 % vs. 40.9 % (P = 0.0012) in placebo vs. vedolizumab groups, respectively.
Those patients achieving a clinical response (reduction in Mayo score of ≥3 points and ≥30 % from baseline plus a decrease in rectal bleeding subscore ≥1 point or absolute rectal bleeding subscore of ≤1 point) in the GEMINI I study after induction therapy at 0 and 2 weeks were randomized to receive vedolizumab 300 mg IV at 4 week intervals or 8 week intervals or placebo for 46 weeks [30, 31]. Three hundred seventy-three patients responded at 6 weeks and were randomized into the maintenance groups. Clinical remission at 52 weeks was seen in 15.9 %, 41.8 %, and 44.8 % in the placebo, 8 weekly, and 4 weekly groups, respectively. Mucosal healing at 52 weeks was seen in 19.8 %, 51.6 %, and 56 %, respectively. Glucocorticoid-free remission was higher in those receiving the drug compared to placebo.
Parikh et al. in 2013 published their long-term clinical experience with vedolizumab in patients with IBD for up to 78 weeks [32]. Thirty-eight patients with UC were randomized to a loading regimen of vedolizumab 2, 6, or 10 mg/kg on days 1, 15, and 43 followed by maintenance every 8 weeks. Fifteen vedolizumab-naïve patients with UC were randomized to vedolizumab in the same dosing/schedule. Seventy-two patients were dosed (53 UC and 19 CD) and 52 (72 %) completed the study. Twenty-one of 53 UC patients achieved clinical response, and 38 of 53 UC patients achieved clinical remission. Mean partial Mayo scores declined from baseline through day 155 both in treatment-naïve patients with UC and rollover patients with UC. No deaths or systemic opportunistic infections were reported, and the adverse event profile was similar to previously observed.
A longer-term phase III trial is ongoing (GEMINI LTS). Patients from the earlier trials will have the option to enter an extended study receiving vedolizumab every 4 weeks for up to 100 weeks. The estimated completion date is March 2016.
In total, 579 patients or volunteers have participated in either phase I or phase II trials with 415 patients having received at least one dose of vedolizumab [22]. There was no significant difference in adverse events between patients receiving vedolizumab and those receiving placebo. The most common adverse events were headache, nausea, exacerbation of UC, abdominal pain, fatigue, and nasopharyngitis. No cases of PML have been reported.
Vedolizumab is a promising new upcoming treatment for UC.
PF-547659
PF-547659 is a monoclonal antibody to MAdCAM-1. By blocking MAdCAM-1, leukocyte migration to the gut mucosa should be altered, thus decreasing the inflammation in UC. See section discussing vedolizumab (Figs. 17.2 and 17.3).
In this first in-human study which was a randomized, double-blind, placebo-controlled trial, 80 patients with active UC received placebo or PF-547659 (0.03–10 mg/kg IV/sc) in single or multiple (three doses every 4 weeks) doses [33–36]. No side effects from the drug were noted. The overall response rates at week 4 were 52 % for patients treated with PF-547659 and 32 % for patients administered placebo. The overall remission rates at week 4 were 13 % for patients treated with PF-547659 and 11 % for patients administered placebo. The overall response rates at week 12 were 42 % for patients treated with PF-547659 and 21 % for patients administered placebo. The overall remission rates at week 12 were 22 % for patients treated with PF-547659 and 0 % for patients administered placebo.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


