Neuroendocrine Tumors of the Gastrointestinal Tract
Matthew H. Kulke
Chandrajit P. Raut
Introduction
Neuroendocrine tumors are generally subcategorized as either carcinoid tumors or pancreatic endocrine tumors. Both tumor types are characterized by variable, but most often indolent, biological behavior, and have, in most cases, characteristic well-differentiated histologic features. Neuroendocrine tumors are typically composed of small cells containing regular, well-rounded nuclei, and are characterized histologically by positive reactions to silver stains and to neuroendocrine markers, including neuron-specific enolase, synaptophysin, and chromogranin (1). The cytoplasm of these cells contains numerous membrane-bound neurosecretory granules, which contain a variety of hormones and biogenic amines. The release of substances such as serotonin, gastrin, glucagon, and insulin into the systemic circulation results in the unique systemic syndromes associated with neuroendocrine tumors.
For patients with localized disease, surgical resection alone is often curative. Patients with metastatic disease, however, often present a therapeutic challenge. Although somatostatin analogs are highly effective in controlling symptoms of hormonal secretion, they are only rarely associated with tumor regression. Selected patients with hepatic metastases may benefit from surgical debulking, embolization, or other ablative therapies. The clinical benefit associated with the administration of systemic agents such as interferon-alpha (IFN-α) or cytotoxic chemotherapy is less clear, and the widespread use of such regimens has been limited by their relatively modest antitumor activity as well as concerns regarding their potential toxicity. The mixed clinical results seen with these agents in neuroendocrine tumors has led to great interest in the development of novel treatment approaches for patients with advanced disease.
Pancreatic Endocrine Tumors
Incidence and Etiology
Pancreatic endocrine tumors are relatively rare, with an estimated incidence of <1 per 100,000 individuals (2). These tumors may arise either sporadically or, less commonly, in patients with multiple endocrine neoplasia type 1 (MEN 1). MEN 1 is most commonly diagnosed in patients with gastrinomas and insulinomas (3,4). MEN 1 is an autosomal dominant syndrome associated with mutations in the MEN 1 tumor suppressor gene, and characterized by multiple neuroendocrine tumors involving the parathyroid and pituitary glands, as well as the pancreas. The protein encoded by MEN 1, menin, has been shown to localize to the nucleus and regulate gene transcription (5). MEN 1 is located on chromosome 11q13, and loss of 11q13 has been demonstrated in both MEN 1-associated pancreatic neuroendocrine tumors and in >50% of sporadic pancreatic neuroendocrine tumors (6). Loss of heterozygosity in regions of chromosomes 22q, 9p, 6q, and 1 has also been observed in pancreatic neuroendocrine tumors, as have point mutations in the DPC4/Smad4 gene (7,8,9,10,11). In rare cases, pancreatic endocrine tumors may be associated with Von Hippel-Lindau disease (12).
Clinical Presentation
The clinical presentations of pancreatic endocrine tumors are diverse and are often related to symptoms of hormonal hypersecretion (Table 48.1). The best characterized of these syndromes are those associated with insulinoma, glucagonoma, VIPoma, and gastrinoma. Additional pancreatic neuroendocrine tumors include somatostatinomas and so-called “nonfunctioning” pancreatic neuroendocrine tumors, which are often associated with high serum levels of pancreatic polypeptide.
Insulinoma
Insulinomas have an estimated annual incidence of one to four cases per million persons (13). These tumors are most common in the fifth decade of life, but diagnoses have ranged from newborn up to the ninth decade (14). Between 8% and 10% of patients with insulinoma also have MEN 1 (13).
The clinical diagnosis of insulinoma is often related to symptoms of hypoglycemia. One of the first described patients with insulinoma is said to have “resembled an acute alcoholic—great motor activity, dancing and taking, squinting and frowning, apparently having hallucinations of sight and hearing, negatavistic, and difficult to control” (15). These symptoms are classic manifestations of hypoglycemia, which can cause both autonomic symptoms and central nervous system dysfunction. Hypoglycemic symptoms have been separated into two categories—adrenergic or neuroglycopenic. Adrenergic symptoms include nervousness, tremulousness, palpitations, anxiety, irritability, diaphoresis, hunger, and pallor, whereas neuroglycopenic symptoms include confusion, headache, personality changes, weakness, blurred vision, dizziness, amnesia, dysarthria, convulsions, and, in severe cases, loss of consciousness (14). The early symptoms of insulinoma can often be
nonspecific, and delays in diagnosis are common. The period from symptom onset to diagnosis may range from 2 weeks to 30 years (13). Indeed, in approximately half of cases, the diagnosis is not made until patients present in a hypoglycemic coma (16).
nonspecific, and delays in diagnosis are common. The period from symptom onset to diagnosis may range from 2 weeks to 30 years (13). Indeed, in approximately half of cases, the diagnosis is not made until patients present in a hypoglycemic coma (16).
Table 48.1 Clinical Presentation of Pancreatic Neuroendocrine Tumors | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
The combination of symptoms of hypoglycemia, inappropriately high insulin levels with associated documented blood glucose levels of <50 mg/dL, and symptom relief with administration of glucose, constitutes Whipple’s triad, first described in 1935 and still useful in the diagnosis of insulinoma (17). The diagnosis of insulinoma can generally be confirmed with the detection of elevated fasting insulin and C-peptide levels in the plasma and the tolbutamide test (14,18).
Because of their association with hypoglycemia, insulinomas are typically small and are often detected before they grow to 2.5 cm (16). In 80% of cases, the tumor is detected without evidence of metastases. Because of their small size, standard imaging modalities such as computed tomography (CT), magnetic resonance imaging (MRI), and angiography are often unsuccessful in identifying the tumor (19,20). Nearly all insulinomas are found in the pancreas, and further imaging studies should focus on this area (14). Endoscopic ultrasound has been reported to be highly sensitive in the preoperative localization of insulinomas (21,22,23). Intraoperative localization techniques, which include both careful palpation of the pancreas and the use of intraoperative ultrasound, remain the most reliable way to localize insulinomas (19).
Dietary modification, together with the administration of diazoxide, is usually successful in the initial management of hypoglycemia due to an insulinoma (24). Isolated insulinomas are generally treated with enucleation; long-term survival following surgery in this patient population exceeds 90% (13). The role of surgical resection in patients with MEN 1 syndrome remains controversial because of the risk of additional tumors within the remaining pancreas and elsewhere. A more aggressive surgical approach, generally with subtotal pancreatectomy, may be undertaken in these patients due to the risk of multiple tumors and a higher rate of recurrence (25,26).
Gastrinomas and Zollinger-Ellison Syndrome
The gastrinoma syndrome, first described by Zollinger and Ellison, is characterized by gastric hypersecretion (27). Gastrin, which is normally secreted by the G cells in the gastric antrum, not only stimulates acid secretion in parietal cells but also acts as a trophic factor, causing parietal cell hyperplasia and an increase in maximal acid output. The profound acid hypersecretion associated with gastrinomas typically causes abdominal pain due to peptic ulcer disease, diarrhea, and reflux esophagitis (28).
Gastrinomas are slightly more common in males than in females, and the median age at presentation is between 40 and 50 years (28,29,30). The diagnosis of gastrinoma is often delayed due to the fact that most patients are initially diagnosed with benign peptic ulcer disease (28,29). A gastrinoma should be suspected in the setting of nonhealing peptic ulcers and a fasting gastrin level of >100 pg/mL (31). The diagnosis can be complicated by the fact that several other conditions can also cause moderate elevations of serum gastrin. The most common of these is concomitant therapy with proton pump inhibitors. In equivocal cases, the documentation of increased basal acid output and a positive secretin provocative test may be necessary to confirm the diagnosis (31,32,33).
Proton pump inhibitors are highly effective in controlling the symptoms associated with gastric hypersecretion (34,35). The localization and surgical resection of gastrinomas often presents a greater challenge. The overwhelming majority of gastrinomas is found in the “gastrinoma triangle,” an area bounded by the cystic and common bile ducts, the duodenum, and the pancreas (36). Within this area, tumors are most commonly found in the duodenum or in surrounding lymph nodes, and less than half are found in the pancreas (29,30,37). Duodenal gastrinomas typically measure <1 cm, making them virtually impossible to detect with standard preoperative imaging studies. Although techniques such as endoscopic ultrasound and somatostatin scintigraphy appear to have some utility in the preoperative evaluation of patients with gastrinoma,
intraoperative palpation and duodenotomy is often still required (29,30,38).
intraoperative palpation and duodenotomy is often still required (29,30,38).
Surgical cure is possible in more than half of the patients with sporadic gastrinomas and localized disease (29). Many gastrinomas in patients with MEN 1 occur in the duodenum, and many of these tumors are not identifiable with preoperative imaging studies. Surgical exploration may therefore be necessary for tumor localization (30,39). In the approximate 25% of patients with Zollinger-Ellison syndrome and MEN 1, surgical resection remains controversial (40). In this patient population, a multiplicity of tumors makes curative resection difficult (41). In cases where an isolated lesion is seen with preoperative imaging studies, however, an attempt at resection may still be warranted (32,42,43). Even after extensive duodenal exploration in such cases, curative resection in patients with MEN 1 may still not be feasible (44). In patients with unresectable or metastatic disease, treatment with somatostatin analogs has been associated with improved control of serum gastrin levels and, in some cases, with tumor stabilization or regression (45).
Glucagonomas
Glucagonomas are among the rarest of the pancreatic endocrine tumors and typically present in the seventh decade of life. Approximately 80% of cases are sporadic, and 20% are associated with MEN 1 (46,47). Although glucagonomas may be associated with diabetes mellitus, clinically significant hyperglycemia occurs in only half of such patients. Patients with glucagonomas are frequently intially diagnosed by a dermatologist, after presenting with necrolytic migratory erythema (Fig. 48.1). This rash, characterized by raised erythematous patches beginning in the perineum and subsequently involving the trunk and extremeties, is found in more than two-thirds of all patients. The etiology of the rash is uncertain; possible causes include amino acid deficiency and zinc deficiency (48,49).
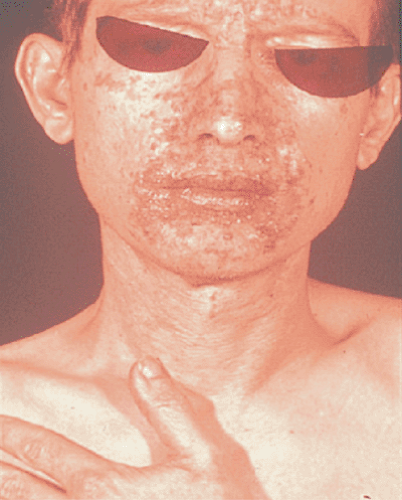 FIGURE 48.1. Necrolytic migratory erythema associated with glucagonoma syndrome (See also color Figure 48.1). |
Plasma glucagon levels, which are <50 pg/mL in normal patients, often exceed 1,000 pg/mL in patients with glucagonomas (50). Somatostatin analogs are generally successful in the initial management of patients with the glucagonoma syndrome (51,52). Patients who are refractory to somatostatin analogs may benefit from the intravenous (IV) infusion of amino acids (46,51). CT is often the only localization study required in patients with glucagonomas, in part because the majority of patients initially present with clearly visible metastatic disease (46,47,53). Surgical resection may be performed with curative intent in the rare patient with localized disease. More often surgery is performed to diminish systemic symptoms in patients with metastases. Perioperative anticoagulation is often considered in such patients due to a high incidence of deep venous thrombosis.
VIPomas
Pancreatic endocrine tumors associated with profound diarrhea, hypokalemia, and achlorhydria were first described by Verner and Morrison in 1958 (54). This syndrome was subsequently found to be due to the ectopic secretion of vasoactive intestinal peptide (VIP) and has been aptly named “pancreatic cholera” (55,56). Like the cholera toxin, VIP causes intracellular elevation of cyclic AMP, resulting in intestinal smooth muscle relaxation, inhibition of electrolyte absorption and profound secretory diarrhea.
VIPomas most often present in the fifth decade of life and have a similar incidence in men and women (57). The diagnosis is generally based on the presence of elevated serum VIP levels, symptoms of diarrhea, and documentation of malignancy. Somatostatin analogs are highly effective in suppressing hormone secretion and in controlling the secretory diarrhea associated with the VIPoma syndrome (58,59). VIPomas are generally >1 cm in size and, in most cases, can be visualized with CT or MRI scan (60,61). Endoscopic ultrasonography or somatostatin scintigraphy may be helpful in localizing smaller tumors. Surgical resection is generally undertaken with curative intent in patients with localized disease, or for the purpose of cytoreduction in patients with metastases (59).
Somatostatinomas and PPomas
Two other types of pancreatic endocrine tumor have been somewhat less well characterized. The first of these, somatostatinomas, may be associated with diabetes, hypochlorhydria, and diarrhea. PPomas are pancreatic neuroendocrine tumors associated with high serum levels of pancreatic polypeptide. Secretion of pancreatic polypeptide is not associated with any clinical syndrome, and these tumors are therefore generally classified as “nonfunctioning” pancreatic endocrine tumors. They are usually first diagnosed when they grow large enough to cause symptoms from tumor bulk (62). Surgical resection is generally curative in patients with early stage disease; unfortunately, because of the clinically silent nature of these tumors, the majority of patients have metastatic disease at the time of diagnosis (63).
Carcinoid Tumors
Incidence and Etiology
The incidence of carcinoid tumors has been estimated to be 1 to 2 per 100,000 population (64). Carcinoid tumors are more common than are pancreatic neuroendocrine tumors: By comparison, the incidence of carcinoid tumors is 8 to 11 times higher than that of insulinomas, and 7 to 26 times higher than that of gastrinomas (65,66,67). An early analysis of 2,837 cases
in the United States using data from the End Results Group (1950–1969) and the Third National Cancer Survey (1969–1971) found that the appendix was the most common site of carcinoid tumors; appendiceal carcinoids were followed in frequency by carcinoids of the rectum, ileum, lungs, and stomach (Table 48.2) (68). A more recent analysis of 5,486 cases identified by the Surveillance, Epidemiology, and End Results (SEER) program of the National Cancer Institute between 1973 and 1991 found a relative increase in the proportion of carcinoids of the lung and stomach, and a decrease in the proportion of appendiceal carcinoids (64). However, these changes in relative incidence are likely due to variations in the detection and reporting of carcinoid tumors. Furthermore, because neuroendocrine tumors often pursue an indolent course, their true incidence may in fact be somewhat higher. The incidence of carcinoid tumors in autopsy series, for example, has been reported to be as high as 8 per 100,000 population, suggesting that a large proportion of patients with carcinoid tumors may in fact die of other, non–tumor-related causes (69).
in the United States using data from the End Results Group (1950–1969) and the Third National Cancer Survey (1969–1971) found that the appendix was the most common site of carcinoid tumors; appendiceal carcinoids were followed in frequency by carcinoids of the rectum, ileum, lungs, and stomach (Table 48.2) (68). A more recent analysis of 5,486 cases identified by the Surveillance, Epidemiology, and End Results (SEER) program of the National Cancer Institute between 1973 and 1991 found a relative increase in the proportion of carcinoids of the lung and stomach, and a decrease in the proportion of appendiceal carcinoids (64). However, these changes in relative incidence are likely due to variations in the detection and reporting of carcinoid tumors. Furthermore, because neuroendocrine tumors often pursue an indolent course, their true incidence may in fact be somewhat higher. The incidence of carcinoid tumors in autopsy series, for example, has been reported to be as high as 8 per 100,000 population, suggesting that a large proportion of patients with carcinoid tumors may in fact die of other, non–tumor-related causes (69).
Table 48.2 Percentage of carcinoid tumors at each site as a proportion of the total number of carcinoid tumors in each study | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
There are no established environmental risk factors for carcinoid tumors, nor has a clear underlying genetic cause for carcinoid tumors been defined. A number of genetic aberrations have been observed in carcinoid tumors; these aberrations appear to differ, depending on site of origin. Although carcinoids are not generally part of the MEN 1 syndrome, mutations or deletions of the MEN 1 gene may be involved in the tumorigenesis of some sporadic carcinoids. Carcinoid tumors arising in the lungs and stomach, for example, have been found to contain chromosomal loss at 11q in approximately one-third of cases (70,71,72). 11q deletions and MEN 1 mutations appear to be less common in carcinoid tumors arising from other sites. One recent study mapped LOH on chromosome 11 using array CGH, and showed that only one of nine midgut NETs had LOH at 11q13, whereas 3 of 9 had LOH at 11q23 (71).
In carcinoid tumors of the small intestine, loss of chromosome 18 has been identified in >50% of cases (73,74). Smad4/DPCr is a gene located on chromosome 18 and often mutates in colon and pancreatic adenocarcinomas; however, no mutations were observed in midgut carcinoids. Other major areas of chromosomal loss in midgut carcinoids include 9p (15%), 11q (13%), and 16q (12%) (10,73,75,76,77).
Clinical Presentation and Management of Localized Carcinoid Tumors
A commonly used classification scheme groups carcinoid tumors according to their presumed derivation from the embryonic gut: foregut (bronchial and gastric), midgut (small intestine and appendiceal), and hindgut (rectal). The clinical presentation and management of these tumors varies, depending on their site of origin (Table 48.3).
Table 48.3 Clinical Presentation of Carcinoid Tumors | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Bronchial Carcinoid Tumors
Bronchial carcinoids comprise approximately 2% of primary lung tumors (78,79). Typical carcinoids, also classified as well-differentiated pulmonary neuroendocrine tumors, usually present in the fifth decade of life (79,80,81). They are most often central in location, causing symptoms of cough, wheezing, hemoptysis, and recurrent postobstructive pneumonia (81,82). Typical pulmonary carcinoid tumors are only rarely associated with the classic carcinoid syndrome; they have, however, been associated with ectopic ACTH secretion, resulting in Cushing syndrome (81,83,84,85).
Approximately one-third of bronchial carcinoids demonstrate “atypical” histologic features (78,82,84,86). Atypical carcinoids are characterized by the presence of frequent mitoses or areas of necrosis. They tend to occur in older individuals, most commonly in the sixth decade of life and, unlike typical carcinoid tumors, are more common in smokers. They also tend to be larger in size than typical carcinoids and are more commonly peripheral in location (87,88).
Whereas typical carcinoid tumors are generally indolent, with metastases reported in <15% of cases, atypical carcinoids pursue an aggressive clinical course, metastasizing to mediastinal lymph nodes in 30% to 50% of cases (80,81,84,85). Accordingly, long-term survival rates for patients with typical carcinoid tumors following surgical resection generally exceed 85%, but are significantly less for patients who undergo resection for atypical carcinoids (78,82,83,84,85,86,88,89,90,91). Histology also influences the choice of surgical procedure: Conservative resection, consisting of wedge or segmental resection, is currently the preferred form of treatment for localized bronchial carcinoid tumors, whereas more aggressive procedures are often chosen for atypical carcinoids (85,88). The use of adjuvant therapy, generally with regimens similar to those used for small cell carcinoma, remains controversial but is at times considered following resection of atypical carcinoid tumors.
Gastric Carcinoid Tumors
Gastric carcinoid tumors comprise <1% of gastric neoplasms (68,92). Gastric carcinoid tumors can be subclassifed into three distinct groups: those associated with chronic atrophic gastritis (type 1), those associated with the Zollinger-Ellison syndrome (type 2), and sporadic gastric carcinoids (type 3) (Table 48.4). Both type 1 and type 2 gastric carcinoids are associated with hypergastrinemia. High levels of gastrin are believed to result in hyperplasia of the enterochromaffin cells in the stomach, ultimately leading to hyperplastic lesions and small, often multiple carcinoid tumors (92). These tumors generally pursue an indolent course and can be resected endoscopically, with subsequent interval follow-up. Patients with larger or recurrent tumors may require more extensive surgical resection. In patients with chronic atrophic gastritis, antrectomy has been used to eliminate the source of gastric production and has been reported to result in tumor regression (93,94). In patients with Zollinger-Ellison syndrome, the use of somatostatin analogs has resulted in tumor regression (95).
Table 48.4 Subtypes of gastric carcinoid tumor | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Between 15% and 25% of gastric carcinoids are sporadic. In contrast to type I and type II carcinoids, these lesions develop in the absence of hypergastrinemia, are usually >1 cm in size, and tend to pursue an aggressive clinical course. Sporadic carcinoid tumors have been associated with an atypical carcinoid syndrome, which is manifested primarily by flushing and believed to be mediated by histamine. The majority of sporadic carcinoid tumors are metastatic at the time of presentation, and death due to disease is frequent. Because of the aggressive nature of these lesions, most are treated with total gastrectomy (96,97).
Small Intestine Carcinoid Tumors
Small bowel carcinoid tumors comprise approximately one-third of small bowel tumors in surgical series (98). Patients with small bowel carcinoids generally present in the sixth or seventh decade of life, most commonly with abdominal pain or small bowel obstruction; these symptoms are commonly misdiagnosed as “irritable bowel syndrome” (98,99,100). Approximately 5% to 7% will present with the carcinoid syndrome, at which time hepatic metastases are usually also present (101,102). The difficulty in diagnosing small bowel carcinoids is compounded by the fact that standard imaging techniques such as CT scan and small bowel barium contrast studies only rarely identify the primary tumor. When detected and surgically removed, they are most frequently located in the distal ileum and are often multicentric, occasionally appearing as dozens of lesions lining the small bowel (Fig. 48.2) (101). Tumor size is an unreliable predictor of metastatic disease, and metastases have been reported even from tumors measuring <0.5 cm (99). The 5-year survival rate is 60% for patients with localized disease and 73% for those with regional metastases, compared to 21% for patients with distant metastases (103).
Mesenteric fibrosis and associated ischemia, caused by a characteristic desmoplastic reaction, is often present in association with small bowel carcinoids. These tumors are also frequently associated with “buckling” or tethering of the intestine due to extensive mesenteric involvement (101,104). Resection of the small bowel primary tumor, together with associated mesenteric metastases, leads to significant reduction in tumor-related symptoms of pain and obstruction, and is therefore
recommended even in patients with known metastatic disease (100).
recommended even in patients with known metastatic disease (100).
 FIGURE 48.2. Multicentric carcinoids of the small intestine (See also color Figure 48.2). |
Appendiceal Carcinoid Tumors
Historically, carcinoid tumors have been considered the most common tumor of the appendix, accounting for >50% of all appendiceal malignancies and discovered in up to 7 of every 1,000 appendectomy specimens (105,106,107). In older surgical series, appendiceal carcinoids were most commonly discovered during incidental appendectomy for other reasons. The most common indication for appendectomy in a 1968 series of 137 appendiceal carcinoid tumors was benign pelvic disease (43%) or gallbladder disease (35%) (103). In recent series, however, the majority of appendiceal carcinoids are discovered during surgery for acute appendicitis (108). The carcinoid tumor itself is believed to be the cause of appendicitis in only a minority of these cases. Approximately two-thirds of appendiceal carcinoid tumors arise in the tip of the appendix, where they are unlikely to cause symptoms of obstruction. Less than 10% arise in the base, where they are more prone to obstruct the appendix, giving rise to acute appendicitis (107).
Patients with appendiceal carcinoid tumors generally present at a relatively young age: The mean age at presentation in the recent SEER database analysis was 49 years, and in older series, it is even younger (103,107). Although the early age at presentation may in part reflect the mean age at appendectomy, other explanations have also been proposed. Some investigators have speculated that the pattern of appendiceal carcinoid tumors parallels the biological behavior of subepithelial neuroendocrine cells, from which appendiceal carcinoid tumors are believed to arise. The density of these cells tends to peak in the third decade of life and subsequently decreases throughout the remainder of life (109,110,111).
Appendiceal carcinoids are more common in women than in men: In the SEER database, the male-to-female ratio was 0.82 (103). The higher incidence of appendiceal carcinoids in women, like the relatively young age at presentation, may be partially attributable to appendectomy patterns. Women are more likely to undergo incidental appendectomy as a result of gynecologic procedures. However, a female preponderance of appendiceal carcinoids has also been reported in children, an observation that cannot be explained by differences in appendectomy rates (112,113,114).
The clinical behavior of appendiceal carcinoid tumors can be predicted based on the size of the tumor. More than 95% of appendiceal carcinoid tumors are <2 cm in diameter (115,116). The incidence of metastatic disease in such patients is extraordinarily low, although rare cases have been reported in the literature (116,117,118,119,120,121). In contrast, approximately one-third of patients with appendiceal carcinoid tumors measuring >2 cm in diameter have either nodal or distant metastases (115). Other criteria such as invasion of the mesoappendix, perineural invasion, tumor location, and histologic pattern have not consistently correlated with the presence of metastatic disease.
The surgical management of appendiceal carcinoids derives from these historical data, and surgical recommendations are based on tumor size. Based on the low incidence of metastases in patients whose tumors measure <2 cm, simple appendectomy is believed to be sufficient in such cases. In contrast, the higher incidence of metastases in patients whose tumors measure >2 cm in diameter has led to the recommendation for complete right hemicolectomy. These recommendations are supported by data from surgical series, one of the largest of which was published by Moertel et al. (115). Among 122 patients undergoing simple appendectomy for tumors measuring <2 cm, none developed disease recurrence, whereas 1 of 12 patients undergoing simple appendectomy for a tumor measuring >2 cm developed a local recurrence. Whether right colectomy decreases the risk of future distant recurrence has not been formally evaluated, and simple appendectomy may at times be appropriate for larger tumors, particularly in older patients or patients with other comorbidities.
Rectal Carcinoid Tumors
Rectal carcinoid tumors comprise 1% to 2% of all rectal tumors, and are most common in the sixth decade of life (64). Approximately 50% are asymptomatic and found on routine endoscopy (122). Symptomatic patients usually present with rectal bleeding, pain, or constipation (122,123). The size of the primary lesion correlates closely with the probability of metastases, which occur in <5% of tumors measuring <1 cm but in the majority of lesions >2 cm (123,124).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


