Chapter 8C Molecular biology of liver carcinogenesis and hepatitis
Overview of Molecular Etiology
Recent advances in molecular genetics have emphasized the multistep process of tumorigenesis. It is evident that cancer is a genetic disease involving aberrant chromosome rearrangements, genetic mutations, and epigenetic silencing of tumor suppressor genes (Farazi & DePinho, 2006). Independent of the etiology, hepatocellular carcinoma (HCC; see Chapter 80) generally develops where sustained hepatocyte turnover occurs in the setting of injury-inflammation-regeneration, which leads to the accumulation of chromosomal aberrations. In this context, monoclonal populations of hepatocytes become preneoplastic and, following additional genomic alterations, change into dysplastic cells and eventually HCC (Thorgeirsson & Grisham, 2002). Accumulated genetic alterations in preneoplastic lesions and HCC result in the activation, as well as inactivation, of many growth factor signal transduction pathways involved in hepatic transformation. It is believed that increased hepatocyte turnover associated with chronic liver injury may be a major feature of hepatic oncogenesis. However, another central question is whether hepatitis viruses, the leading cause of HCC worldwide, directly contribute to the development of this disease. Accumulating evidence suggests that chronic hepatitis B (HBV) and hepatitis C virus (HCV) infection play a direct role in the molecular pathogenesis of HCC through specific viral–cellular protein interactions (Branda & Wands, 2006; see Chapter 64).
Epidemiology
Hepatocellular carcinoma is the fifth most common cancer in men and the eighth most common in women worldwide. It is estimated that more than 700,000 new patients with HCC were diagnosed in 2007 (Garcia et al, 2007). The 5-year survival rate is less than 11% in developed countries, and the United States has a survival rate of only 8.9%, making HCC the second most fatal tumor after pancreatic cancer (Hertl & Cosimi, 2005; Garcia et al, 2007). Presumably because of its poor prognosis, HCC is the third leading cause of cancer death in men and the sixth among women in the world. It is estimated that about 680,000 individuals worldwide died from this disease in 2007 (Garcia et al, 2007).
Primary liver cancers consist of numbers of histologically distinct types of tumors that arise from hepatocytes, biliary epithelial cells, and fibroblasts. The most common is HCC, which accounts for 70% to 85% of all hepatic tumors (Perez et al, 2006). Approximately 80% of HCC worldwide is caused by chronic infection with HBV or HCV or both. The HCC burden is unevenly distributed worldwide; areas where tumors are most prevalent include West and Central Africa and East and Southeast Asia, with China alone accounting for more than 50% of the world cases (El-Serag & Rudolph, 2007). It is noteworthy that HBV is highly endemic in these areas, with the exception of Japan, where HCV is more prevalent. Oceania, North and South America, and Northern and Eastern Europe are low-rate areas. Trends in HCC incidence are likely to be different in regions of high and low persistence of HBV and HCV infection (El-Serag & Rudolph, 2007).
Comparative studies performed between 1977 and 1982 and between 1993 and 1997 show that the incidence of HCC in Hong Kong, Shanghai, Singapore, and Japan has begun to decrease (Parkin et al, 2002). The fall in incidence is apparently due to vaccination against HBV, which has been accomplished in over 80% of newborns (Chang et al, 2009), because chronic HBV infection in those countries is usually acquired through mother-to-newborn or sibling-to-sibling transmission at a young age. In contrast, the incidence of HCC has rapidly increased in some countries, such as Australia, the United States, and the United Kingdom, as a result of chronic HCV infection. For example, HCC is the fastest growing cause of cancer-related deaths in the United States, where the annual incidence increased from 1.3 per 100,000 for the years 1978 through 1980 to 3.3 per 100,000 for 1999 through 2001 (El-Serag et al, 2003). Reasons for this increased incidence are not entirely clear but may reflect a greater prevalence and role of persistent HCV infection (McGlynn et al, 2001).
Age-specific rates of HCC peak at 75 years and older in most regions of the world (El-Serag & Rudolph, 2007). With the exception of Africa, the peak incidence in women occurs 5 years later than that found in men. In the United States, recent trends have revealed a peak incidence shifting toward a relatively younger age group (El-Serag & Rudolph, 2007).
Significant gender and ethnic variation in incidence, as well as mortality from HCC, has also been found; male rates are more than double that of females (Perz et al, 2006). The most likely explanation for gender variation is that men have more risk factors, such as exposure to hepatitis virus infection, excessive alcohol intake, smoking, and increased iron stores in the liver (El-Serag & Rudolph, 2007). In addition, androgens may accelerate the progression of HCC (Ma et al, 2008). The incidence of HCC also varies with race and ethnicity in the same area. In the United States, the incidence and subsequent mortality rates from HCC are two times greater in Asians than African Americans, which are two times greater than those found in whites (El-Serag & Rudolph, 2007). These variations are explained in part by the accumulation of major risk factors in each ethnic group.
Risk Factors
Unlike most malignancies, HCC has well-established extrinsic risk factors that account for at least 80% of tumors, namely chronic infection with HBV or HBC. Key epidemiologic aspects of HBV- and HCV-induced HCC are summarized in Table 8C.1. Chronic HBV infection is the leading cause of HCC, and it has been estimated that there are 350 million HBV carriers, which account for 6% of the global population. About 59% of HCC patients in developing countries and 23% of HCC patients in developed countries are chronically infected with HBV (Garcia et al, 2007). Reported relative risks of HCC among HBV carriers range between 5-fold and 15-fold compared with the general uninfected population (El-Serag & Rudolph, 2007). The 5-year cumulative incidence rates of HCC from HBV-related cirrhosis are 15% in highly endemic areas and 10% in Europe (Fattovich et al, 2004). In addition, approximately 70% to 90% of HBV-related HCC develops in patients with cirrhosis.
Table 8C.1 Comparison of Epidemiologic Features Between HBV- and HCV-Induced HCC
| HBV | HCV | |
|---|---|---|
| Virus carriers (% of global population) | 350 million (6%) | 170 million (3%) |
| Highly prevalent areas | Southeast Asia, China, sub-Saharan Africa, Alaska, Peru, northwest Brazil | East Asia, Southeast Asia, Africa, Bolivia, Brazil |
| Relative risk of HCC | 5-fold to 15-fold | 17-fold |
| 5-Year cumulative incidence rates of HCC from cirrhosis | 10% (Europe) 15% (Singapore and Taiwan) | 17% (Europe and United States) 30% ( Japan) |
HBV, Hepatitis B virus; HCV, hepatitis C virus; HCC, hepatocellular carcinoma.
Chronic HCV infection is the second leading cause of HCC. The estimated number of HCV carriers worldwide is 170 million, which accounts for 3% of the global population. Approximately 33% of HCC tumors in developing countries and 20% of HCC in developed countries are attributable to persistent HCV infection (Garcia et al, 2007). According to a meta-analysis of case-control studies, HCC risk was increased 17-fold in HCV-infected persons compared with the HCV-negative population (Donato et al, 2002). The 5-year cumulative incidence of HCC with HCV-related cirrhosis in developed countries is 17%, with an exception for Japan, where the 5-year cumulative incidence is 30% (Fattovich et al, 2004). The high incidence of HCV-related HCC in Japan may be due to the prevalence of HCV genotype 1b. Aflatoxin B1 (AFB1) is produced by Aspergillus flavus and related fungi that contaminate corn, rice, and peanuts in China and sub-Saharan Africa. High rates of dietary exposure to AFB1 increase the risk of HCC 4-fold. When people with chronic HBV infection are exposed to AFB1, the relative risk for HCC dramatically increases to about 60-fold (Kew, 2003). This synergistic effect between AFB1 exposure and chronic HBV infection is an important observation because in some regions of the world, AFB1 exposure and chronic HBV infection rates are high.
Excessive ethanol consumption (>50 to 70 g/day) is another well-defined risk factor for HCC. In the United States, ethanol-induced HCC accounted for more than 20% of HCC patients between 1996 and 1999 (Davila et al, 2004). The 5-year cumulative HCC incidence in alcoholic cirrhosis without HBV and HCV infection is 8% (Fattovich et al, 2004); however, it is unlikely that ethanol itself has a direct carcinogenic effect. Rather, excessive ethanol ingestion indirectly affects hepatocarcinogenesis through the promotion of cirrhosis. Indeed, more than 80% of HCC tumors found in alcoholics develop in the background of a cirrhotic liver. One case-control study observed a strong synergistic effect between heavy alcohol consumption and chronic HCV infection. The relative risk of HCC attributable to heavy alcohol consumption alone was only 2.4-fold, whereas in combination with chronic HCV infection, it increased to 50-fold (Hassan et al, 2002).
Growing evidence now suggests that metabolic dysfunction—including obesity, diabetes, and nonalcoholic fatty liver disease (NAFLD)—are important risk factors for HCC, especially in developed countries. Several large cohort studies reveal that obese men have 2-fold to 5-fold higher rates of HCC. The association of HCC with obesity in women is controversial. Other investigations suggest that diabetes is a moderately strong risk factor for HCC (El-Serag & Rudolph, 2007). Nonalcoholic steatohepatitis (NASH) is strongly associated with obesity and diabetes; therefore NASH may be a risk factor for HCC (see Chapter 65). Epidemiologic studies have searched for a direct correlation between NASH and HCC, but the results have not been sufficiently convincing to draw a firm conclusion.
Other risk factors for HCC include hemochromatosis and hepatic porphyria. It should be noted that daily coffee intake reduces the incidence of HCC between 25% and 75% in the general population in a dose-dependent manner (Inoue et al, 2005). This effect may be attributable to the inhibition of TGF-β signaling by methylxanthine caffeine, which reduces liver fibrosis (Gressner, 2009).
Genetic and Epigenetic Alterations
Chronic inflammation accompanied by sustained cycles of injury and regeneration of hepatocytes over 20 to 40 years promotes the development of liver fibrosis, cirrhosis, and eventually HCC (Fig. 8C.1A; see Chapter 6). Pathologically, HCC occurs early within cirrhotic nodules, which can form in areas of adenomatous hyperplasia or dysplasia. These cells eventually become more atypical, and the malignant transformation process becomes complete (Theise et al, 2002). Hepatocellular carcinoma is like other malignancies and represents a DNA disease with accumulation of many alterations in oncogenes and tumor suppressor genes. The accumulation of genetic aberrations that induces cellular transformation may take 20 to 40 years, suggesting that liver carcinogenesis involves a multistep process. Indeed, the number of genetic alterations in HCC correlates with the histologic differentiation of the tumors. For example, loss of heterozygosity (LOH) of chromosomes 16q, 17p, 13q, and TP53 tumor suppressor genes was principally found in poorly differentiated HCC, which suggests these chromosomal alterations and mutations tend to occur at a late stage of tumor development (Laurent-Puig & Zucman-Rossi, 2006). In contrast, β-catenin mutations were associated with well-differentiated HCC and a more favorable prognosis, indicating that these mutations may occur at an early developmental stage (Mao et al, 2001).
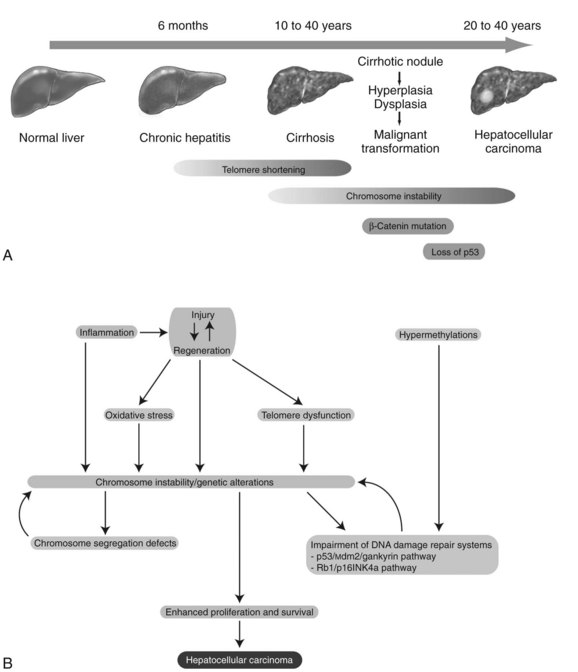
Cirrhosis may represent end-stage liver disease as a result of persistent HBV or HCV infection. Because 80% to 90% of HCC tumors originate from cirrhotic liver, it is evident that continuous rounds of cellular injury followed by regeneration in the milieu of chronic inflammation fundamentally contributes to the oncogenic processes. The sustained cycles of injury and repair increase the chance of genomic alteration. Furthermore, the host inflammatory response to viral infection, including activation of stellate cells, causes the release of proinflammatory cytokines, which accelerate hepatic carcinogenesis by augmenting oxidative stress and DNA damage (Bataller & Brenner, 2005; Giannelli et al, 2005; Ogata et al, 2006).
Continuous rounds of this process in the presence of inflammation not only increase the chance of genomic alterations but also produce chromosome instability. For example, hyperploidy has been observed in 43% of dysplastic peritumoral regions and in about 50% of HCC tumors (Laurent-Puig & Zucman-Rossi, 2006). Molecular mechanisms underlying such genomic instability include telomerase dysfunction, defective segregation of chromosomes, and an impaired DNA damage response (Fig. 8C.1B).
Telomere shortening is a key feature of chronic liver disease that allows sustained proliferation of hepatocytes (Urabe et al, 1996). In human HCC, telomere shortening has been shown to have a positive correlation with increased chromosome instability—chromosomal gains, losses, and translocations—by promoting chromosomal fusions (Plentz et al, 2004). A study of telomerase-deficient mice reveals that telomere dysfunction initiates tumor formation (Farazi et al, 2003). It is noteworthy that 90% of human HCC shows robust activation of telomerase (Lee et al, 2004; Nagao et al, 1999; Shimojima et al, 2004). In some HBV-induced HCC tumors, the viral genome was found to be integrated into the TERT locus, which results in increased expression of telomerase (Ferber et al, 2003; Murakami et al, 2005). Other findings related to telomerase biology indicate amplification of telomerase RNA component gene (TERC) mRNA and allelic loss of chromosome 10p, where a putative telomerase inhibitor resides (Nishimoto et al, 2001; Takeo et al, 2001). Such telomerase reactivation and telomere shortening in HCC cells may be explained in part from investigations performed in TERC-knockout mice (Farazi et al, 2003). This study indicates that telomerase reactivation occurred and appeared to be necessary for late-stage tumor progression; thus, the reactivated telomerase enzyme maintains the shortened telomere length in these tumor cells and prevents them from undergoing apoptosis.
Dysregulation of genes involved in chromosome segregation results in altered copy numbers during cell division. Aneuploidy has been frequently found in HCC, suggesting dysfunction of the segregation machinery. Indeed, aurora kinase A, a protein required for proper chromosome segregation, was shown to be abnormal (Smith et al, 2003). Furthermore, the spindle-assembly checkpoint is defective in HCC cell lines (Saeki et al, 2002). These cytogenetic dysfunctions may lead to the aberrant segregation of chromosomes in human HCC.
DNA damage-response pathways are safeguards that regulate cell-cycle checkpoints and prevent DNA-damaged cells from further proliferation. Several studies report that the functions of key regulatory molecules, including p53, Mdm2, Rb1, p16INK4a, and gankyrin, were impaired in human HCC. The p53 protein, encoded by the TP53 gene, is a master molecule that maintains genome integrity by inducing cell-cycle arrest followed by activation of DNA repair systems. When excessive DNA damage occurs, p53 initiates apoptosis. TP53 missense mutations in codon 249 (R249S) was the main cause of AFB1-induced liver cancer (Bressac et al, 1991). This specific TP53 mutation was found in more than 50% of AFB1-related HCC; other mutations of TP53 are found in 20% to 40% of HCC without molecular evidence of AFB1 exposure (El-Serag & Rudolph, 2007).
The Rb1 pathway also regulates cell cycle checkpoints. The loss of an RB1 locus, located on chromosome 13q14, has been frequently observed in human HCC; the second allele may then be inactivated by an epigenetic mechanism, such as hypermethylation of promoter sequences to promote cell proliferation (Zhang et al, 1994; Lin et al, 1996). Cellular levels of p53 and Rb1 proteins are low due to rapid degradation. In this regard, Mdm2 an E3 ubiquitin ligase, was shown to be a key regulator of ubiquitin-proteasome degradation of these tumor suppressor proteins. Strikingly, gankyrin, which promotes such protein degradation by Mdm2, was overexpressed in 100% (n = 34) of human HCC (Higashitsuji et al, 2000, 2005). The CDKN2A gene encodes for two splice variant products, including p16INK4a and p14ARF, which are components of the p53 and Rb1 signaling pathways. The expression of the CDKN2A gene was suppressed in 30% to 70% of human HCC as a result of methylation of the promoter region (Jin et al, 2000; Liew et al, 1999; Matsuda et al, 1999; Weihrauch et al, 2001). LOH of chromosome 9p where CDKN2A is located was found in 15% to 20% of tumors (Boige et al, 1997; Laurent-Puig et al, 2001; Nagai et al, 1997). Interestingly, CDKN2A deletion rarely occurs in HCC when TP53 is also mutated (Tannapfel et al, 2001). Taken together, impairment of the p53 and Rb1 pathways is a common genetic feature of HCC. In addition, chromosome instability caused by telomere shortening, as well as defects in chromosomal segregation, contributes to the molecular abnormalities often observed in this disease.
As a result of chromosome instability and impaired DNA repair systems, chromosome gains and losses and gene mutations are frequently found in human tumors, as shown in Table 8C.2. Numerous genetic studies performed during hepatocarcinogenesis provide an overview of chromosomal aberrations by using karyotypic analysis, LOH mapping, and comparative genomic hybridization (CGH). To summarize, chromosome 17p (TP53), 8p (PINX1), 16p (AXIN1), 16q, 4q (NAP1L5), 9p (CDKN2A), 13q (RB1, BRCA2), 1p, and 6q (IGF2R) were often deleted. Chromosomes 1q (HDGF), 7q, 8q (MYC), and 17q (ERBB2) were the most frequent to shown gain of genetic material (Laurent-Puig & Zucman-Rossi, 2006). Some chromosome gains and losses have been reported to be specific to etiologic factors such as chronic HBV and HCV infection. Notably, gains of chromosome 8q and 20q and loss of chromosome 4q have been found in HBV-induced HCC without cirrhosis, indicating that these chromosomal aberrations may enhance hepatocyte transformation (Feitelson et al, 2002; Wong et al, 1999).

It is evident that the expression and function of oncogenes and tumor suppressor genes are affected by copy number because of chromosomal gains and losses and by point mutations in the genes. However, recent studies have revealed that epigenetic mechanisms—such as DNA methylation and short, noncoding RNA (21 to 23 nucleotides) species, or micro RNA (miRNA)—also contribute to aberrant expression of oncogenes and tumor suppressor genes. In human HCC, aberrant DNA methylation patterns have been detected (Kanai et al, 1999; Thorgeirsson & Grisham, 2002; Yu et al, 2003). More important, hypermethylation has been observed at the earliest stages of HCC development, and the extent of hypermethylation tends to increase with tumor progression (Lee et al, 2003). Specific gene targets for hypermethylation include CDKN2A, PTGS2, CDH1, PYCARD, and DLC1. Among these genetic elements, it has been shown that CDKN2A and PTGS2 expression were directly affected by methylation using human HCC cell lines (Liew et al, 1999; Matsuda et al, 1999; Murata et al, 2004).
In addition, miRNA contributes to messenger (mRNA) instability by hybridizing with its complementary target sequence, followed by mRNA degradation, so that a protein cannot be generated. Several studies reveal aberrant expression of some miRNAs in human HCC compared with the adjacent, nontumorous counterpart. For example, miR-21 was expressed in human HCC; it targets the PTEN tumor suppressor gene (Meng et al, 2007). Another example was overexpression of miR-122, which targets the cyclin G1 cell-cycle regulator (Gramantieri et al, 2007). More recently, evidence was presented that links aberrant expression of miRNA to the multistep process of hepatocarcinogenesis. The expression of miR-26a was diminished in murine and human tumors, resulting in enhanced activity of cyclin D2 and E2 to promote cell proliferation. Moreover, when exogenous miR-26a was overexpressed in mice prone to form multiple HCCs, substantial protection from disease progression was observed, indicating a possible therapeutic approach for this disease (Kota et al, 2009). These findings indicate that epigenetic and posttranscriptional regulation of gene expression plays an important role in hepatic oncogenesis.
Signal Transduction Pathways
Genetic alterations of oncogenes and tumor suppressor genes impinge on a wide variety of signal transduction pathways involved in proliferation and tumor cell viability. Although the spectrum of affected signal transduction pathways in HCC cells is more heterogeneous compared with that of affected signals in other tumor types, key pathways are commonly dysregulated in human HCC, such as the Wnt/β-catenin, ErbB/ERK/PI3K, and IGF/IRS/ERK/PI3K cascades (Fig. 8C.2).
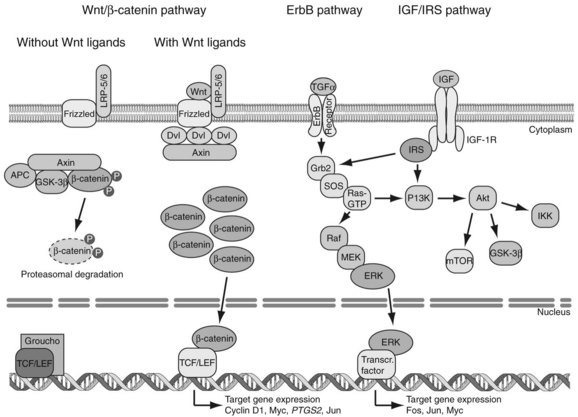
The Wnt/β-catenin pathway regulates cell proliferation, motility, and differentiation. Wnt proteins are ligands that bind to Frizzled cell-surface receptors to stabilize β-catenin in the cytoplasm, followed by translocation to the nucleus, where it upregulates Wnt-responsive genes (Thompson & Monga, 2007
Related posts:
 Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
 Nonhepatic surgery in the cirrhotic patient
Nonhepatic surgery in the cirrhotic patient
 Liver transplantation for cholangiocarcinoma and other neoplastic diseases
Liver transplantation for cholangiocarcinoma and other neoplastic diseases
 Distal splenorenal shunt
Distal splenorenal shunt
 Interventional endoscopy: Technical aspects
Interventional endoscopy: Technical aspects
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


