Antimicrobial susceptibility testing is needed to adapt Helicobacter pylori treatment to obtain the best results. Beside the standard phenotypic methods, molecular methods are increasingly used. The value of these molecular tests is that they are quick, independent of the transport conditions, easy to standardize, and commercial kits are available. In this article, these methods are reviewed, focusing on the determination of H pylori resistance to macrolides and fluoroquinolones, and mentioning also the methods used for tetracycline and rifampin.
Key points
- •
Detection of antimicrobial resistance of Helicobacter pylori is important to tailor the treatment and obtain the best outcome of eradication.
- •
Molecular methods that detect mutations in genes relevant to antimicrobial resistance can be applied, especially for the most important antibiotic (ie, clarithromycin).
- •
Numerous molecular methods have been proposed to detect the main 3 mutations associated with clarithromycin resistance of H pylori , the most commonly used being real-time polymerase chain reaction protocols.
- •
The correlation between molecular detection of resistance via mutations and antimicrobial susceptibility testing by Etest is not perfect, because the former is better for detecting heteroresistance, but which method correlates the best with eradication is not known.
- •
Molecular methods can also be applied to detect H pylori resistance to fluoroquinolones, tetracycline, and rifampin, although they are not so commonly used.
- •
The advantage of molecular methods is their rapidity, lack of stringent transport conditions, and standardization.
- •
Their limit is that they cannot be used for all antibiotics and they do not detect resistance caused by mutations other than those already known or other resistance mechanisms.
There are several reasons for failure of the treatments aiming to eradicate Helicobacter pylori . They include a poor compliance to the regimen and a high gastric acidity, which is not overcome by the recommended dose of proton pump inhibitor (PPI) that increases the minimal inhibitory concentration (MIC) of the antibiotics used. In the past, different conditions, such as an important bacterial load, infection by CagA (cytotoxin-associated gene A)-positive versus CagA-negative H pylori strains, and the presence of intracellular bacteria and some immunologic deficiencies have been suggested to influence eradication but seem less important when susceptibility and compliance are taken into consideration.
H pylori may become resistant to all the antibiotics used for eradication in the various regimens proposed, essentially according to the same mechanism (ie, acquisition of point mutations). Point mutations occur by chance, and increase the MIC of the bacteria. Those organisms with point mutations are then selected by the corresponding antibiotics when prescribed. Another mechanism that sometimes occurs is an efflux mechanism of resistance (ie, efflux pumps, which tend to eliminate the antibiotic having penetrated into the bacterial cell).
Acquisition of resistance in H pylori is important essentially for macrolides (clarithromycin) and fluoroquinolones (levofloxacin). It rarely occurs for β-lactams (amoxicillin), tetracyclines, and for rifampin (rifabutin). To the contrary, although they seem to be frequent for 5-nitroimidazoles (metronidazole), they can be overcome in vivo.
As for any infection, it seems crucial to detect H pylori resistance before prescribing a treatment, the efficacy of which would be jeopardized by the presence of resistant organisms.
The standard detection method consists of performing an antibiogram, usually or MIC determination, using Etest. Although this procedure has the advantage of offering testing of all of the antibiotics of interest, it also has some drawbacks. It requires living organisms, and culturing H pylori is sometimes challenging because of the special transport conditions necessary for gastric biopsies, as well as special care in the laboratory; several days are necessary for primary culture and then performing the antibiogram. For these reasons, alternative methods to this phenotypic approach have been proposed, including various molecular approaches.
The aim of this article is to review these methods, focusing on the determination of H pylori resistance to macrolides and fluoroquinolones, which are the most important, and mentioning also the methods used for tetracycline and rifampin.
Molecular determination of Helicobacter pylori resistance to macrolides
Mechanisms
Macrolides target the 23S ribosomal RNA (rRNA). There are in particular 2 nucleotide positions at the domain V level of the peptidyl transferase loop, which can lead to resistant organisms, because they induce a change in the ribosome conformation and decrease macrolide binding. These positions are 2142 and 2143. A transition can be found at both positions, whereas a transversion is found only at the former ( Fig. 1 ). Other mutations that could theoretically occur are not found in nature, possibly because they lead to nonviable organisms. Some reports of other mutations associated with clarithromycin resistance have been made but could not be confirmed.
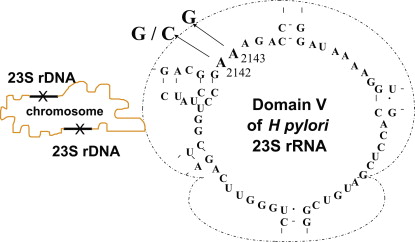
However, a recent study questions this dogma. Comparing phenotypic and genotypic resistance to clarithromycin, De Francesco and colleagues found a high rate of discrepancy. Of 42 clarithromycin-resistant strains, only 23 harbored the 3 known mutations, whereas 19 did not. These investigators identified the following mutations in 14 of 19 cases: A2115G, G2141A, and A2144T.
Confirmation of such findings has not yet been made. In our recent experience (2014), there were only 3 discrepancies between genotypic and phenotypic methods out of 400 strains tested (Mégraud, 2015).
Others mechanisms that could be involved in clarithromycin resistance concern efflux pumps, as was found in campylobacters.
Methods
There are various molecular methods to detect these mutations, essentially polymerase chain reaction (PCR)-based methods as well as a non-PCR-based method, fluorescent in situ hybridization (FISH).
One of the most efficient methods is real-time PCR. This method is described first. The other techniques are also reviewed.
Real-time polymerase chain reaction for detection of Helicobacter pylori resistance to macrolides
The beauty of this method is that it is first able to detect the presence of H pylori with a better sensitivity than other methods, including culture. In our experience, 3% to 5% of true positives detected by PCR cannot be cultured probably because of preanalytical problems.
Principle
The first step consists of designing primers specific for H pylori on the 23S rRNA gene used as the target gene, on both sides of the mutation site. The second step is to design probes inside the fragment to be amplified: a 3′ anchor probe labeled with a fluorochrome (eg, fluorescein) and a 5′ sensor probe labeled with another fluorochrome (eg, LC-Red640), which must be located close to the other (3 bases upstream) to allow an energy transfer from the former to the latter. This is the principle of so-called fluorescence resonance energy transfer (FRET), performed in a LightCycler thermocycler (Roche Diagnostics, Neuilly sur Seine, France), which allows the amplicon formation to be followed in real time.
If the 23S rRNA gene of H pylori is present in the mixture, a curve is obtained after 35 cycles, allowing the identification of an H pylori –positive specimen.
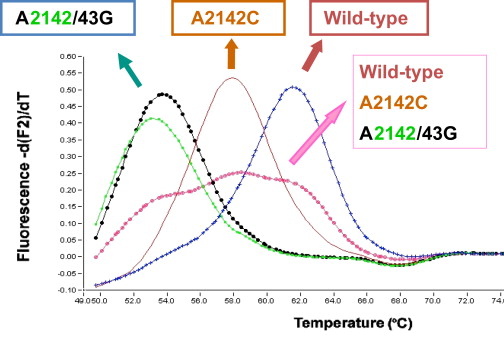
Then, a melting curve analysis (MCA) is performed (ie, the temperature of the mixture is increased to determine the temperature at which dissociation of the double amplicon strands occurs). In the case of a wild-type, the melting temperature is the highest (62°C), whereas in the case of a mismatch, the melting temperature is lower: 58°C for the transversion and 53°C to 54°C for the transition ( Fig. 2 ). This approach was also performed by Matsumura and colleagues with different primers and probes.
Historically, this method was first proposed using SYBR green, a fluorophore specific for double-stranded DNA and used as a quencher, which transfers its energy to a second fluorophore Cy5 fixed on a probe specific to H pylori .
Advantages of this procedure
- •
It is not necessary to have viable organisms, so the transport conditions are not as strict as for culture, because DNA remains unaltered even at ambient temperature during long periods.
- •
The procedure can be performed within a few hours. DNA must be extracted: this can be done in various ways, including the use of commercial kits, and it can also be automatized. Then, the amplification reaction is performed within 2 hours.
- •
Mixed populations made of resistant and susceptible bacteria can be better detected than by traditional-based culture methods.
- •
There are commercially available PCR kits that offer a standardized procedure: ClariRes Assay (Ingenetix, Vienna, Austria), MutaReal (Immundiagnostic, Bensheim, Germany). Another interesting point is that this methodology can be applied not only to fresh biopsy specimens but also to archival material (eg, fixed material on histologic preparations), as well as on other specimens in which culture is seldom positive (ie, stool specimens). However, the accuracy of the results of H pylori detection in stools is still controversial. The amount of H pylori DNA in stools is not important, and inhibitors of the Taq polymerase may decrease the sensitivity of the method unless a long DNA extraction procedure is performed.
A variation of the method proposed by Lascols and colleagues consists of a quantitative detection of H pylori in gastric biopsy specimens followed, in the event of a positive result, by the performance of another hybridization with a different biprobe, followed by an MCA.
Variants of the method
The TaqMan format
For each of the 3 known mutations (A2142C, A2142G, and A2143G) and the wild-type of the 23S rRNA gene, 4 TaqMan-MGB (Minor Groove Binder) probes are designed. A first probe having the fluorochrome VIC allows the detection of the wild-type, a second labeled with fluorochrome FAM detects the mutated form A2142C, a third FAM probe the A2142G mutation, and a fourth FAM probe the A2143G mutation. It is necessary to perform several amplifications for each specimen and test the corresponding probes.
The fluorescence emitted by the activated fluorochrome hydrolyzed probe is then detected.
Advantages and limitations
The absence of melting curve requires different amplifications to be performed for each sample and the use of 4 TaqMan probes, which increases the cost.
The scorpion format
An alternative method described by Burucoa and colleagues is based on a single-vessel multiplex real-time PCR that detects H pylori infection and the wild-type sequence and the 3 mutations conferring clarithromycin resistance using allele-specific scorpion primers directly on biopsy specimens. The scorpion primers combine a primer and a probe in a single molecule and are able to distinguish between single nucleotide polymorphisms. Fluorescent signals produced when the probes are annealed are read in 4 channels by a SmartCycler thermocycler (Cepheid, Sunnyval, CA, USA).
Multiplex polymerase chain reaction followed by strip hybridization
The principle is the same as for real-time PCR (ie, the mixture contains primers to specifically amplify H pylori and others targeting the 23S rRNA gene). Once the PCR is carried out, there is a second step of DNA strip hybridization. Strips coated with different oligonucleotides (DNA probes) are commercially available. The probes are designed to hybridize with the sequences of the wild-type alleles or the mutated alleles.
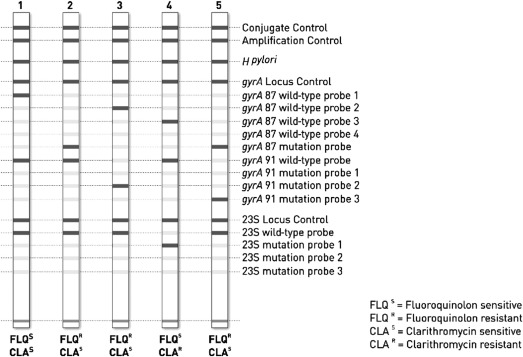
To assess positive and negative bands, the strips are pasted to an evaluation sheet after hybridization, and a template is aligned with the conjugate control band of the respective strip. Control bands of the conjugate control and amplification control should appear positive ( Fig. 3 ). Using the commercially available GenoType HelicoDR (Hain LifeScience, Nehren, Germany) a concordance score of 0.96 was found with real-time PCR for clarithromycin resistance. The sensitivity and specificity in the published studies are presented in Table 1 .
| Country | Specimen | N | Macrolide | Fluoroquinolone | Reference | ||
|---|---|---|---|---|---|---|---|
| Sensitivity (%) | Specificity (%) | Sensitivity (%) | Specificity (%) | ||||
| France | Gastric biopsies | 105 | 97.9 | 100 | 92.3 | 97.4 | Cambau et al, 2009 |
| Strains | 92 | 89.6 | 97.7 | 82.9 | 100 | — | |
| Belgium | Gastric biopsies | 128 | 100 | 86.2 | 82.6 | 95.1 | Miendje Deyi et al, 2011 |
| Korea | Gastric biopsies | 101 | 94.9 | 87.1 | 98.2 | 80 | Lee et al, 2005 |
| South Africa | Strains | 78 | 98 | 100 | 89 | 93 | Tanih & Ndip, 2013 |
A similar prototype named line probe assay was developed earlier, in which oligonucleotide probes were immobilized on a strip (Innogenetics now Fujirebio Europe, Gent, Belgium).
Advantages and limits
As with real-time PCR, this method does not require specific transport conditions and can be performed rapidly. However, it is technically more demanding, because the procedures are not automated. Furthermore, the result is based on human interpretation, and it is sometimes difficult to identify weak bands present on the strip.
Because standard PCR is performed, a risk of amplicon contamination exists, and therefore, strict pre and post PCR conditions are imperative.
On the other hand, this method allows the detection of other resistance mutations present, especially those related to fluoroquinolones, as discussed in the corresponding section.
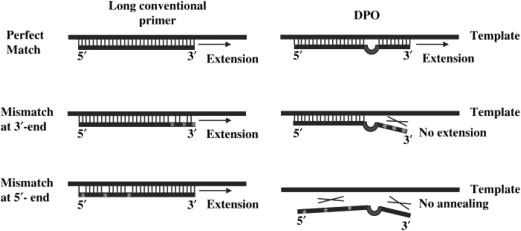
Dual priming oligonucleotide–polymerase chain reaction
This is a multiplex PCR assay that increases specificity and sensitivity of detection compared with conventional PCR, by blocking nonspecific binding sites and then eliminating imperfect primer annealing ( Fig. 4 ). The structure of the dual priming oligonucleotide (DPO) primers is basically different from that of conventional primers. The primer is divided into 2 parts by a 5 polydeoxyinosine linker, which allows a more specific hybridization at temperatures between 55°C and 65°C. This linker forms a bubblelike structure, which itself is not involved in priming but delineates the boundary between the 2 parts. It then generates 2 recognition reactions of the primer on the target sequence. According to the manufacturer, the 5′ end (approximately 20 bases) binds preferentially to the matrix and initiates stable annealing acting as a stabilizer. The 3′ end is shorter (approximately 10 bases) and binds afterward to the target site, but only if the first step has taken place without a mismatch. The 3′ end determines a target-specific extension and acts as a determiner. Therefore, although the longer 5′ segment binds to a nontarget site, the shorter segment resists nonspecific extension. The short 3′ portion alone fails to make a priming at an annealing temperature. The latter also binds preferentially to the target and avoids nonspecific binding. This PCR can be performed in any conventional thermocycler, and a kit is commercially available, the Seeplex ClaR- H. pylori ACE detection kit (Seegene, Seoul, Korea).
The performances of the DPO-PCR have been evaluated in several studies. In the study of Siffre and colleagues, the sensitivity and specificity versus culture were 97.7% and 83.1%, respectively. Like real-time FRET-PCR, DPO-PCR detected 14 positive samples, which were negative by culture. Both methods were concordant in 95% of cases with regard to clarithromycin susceptibility.
Other polymerase chain reaction–based assays
Nested polymerase chain reaction followed by sequencing or standard polymerase chain reaction followed by sequencing or restriction length polymorphism or colorimetric detection of the amplicons
In the early days, the method to detect clarithromycin resistance consisted of a standard PCR targeting the 23S RNA gene. To detect the point mutation, amplicon sequencing can be performed but, for laboratories lacking a sequencing machine, other approaches were proposed. A popular one was to perform a restriction on the DNA fragment. This method is based on the fact that mutations show restriction sites within the amplicon, which are recognized by Bsa I for the A2142G mutation and Bsb I for the A2143G mutation, leading to the presence of 2 bands on the gel instead of 1. Later, a third enzyme ( Bce AI) was proposed to detect the A2142C mutation. This method is interesting but has recently been replaced by more rapid real-time PCR-based methods, furthermore eliminating the risk of amplicon contamination.
Another alternative is to detect mutations by a colorimetric hybridization assay in liquid phase. For this purpose, 5′ biotinylated probes were developed to be specific for each genotype (wild-type and the 3 mutants A2143C, A2143G, A2144G). The cutoffs were determined by receiving operating characteristic curves, and a very high accuracy was obtained.
The discrimination between allelic sequence variants can also be performed by 2 colorimetric methods: (1) the oligonucleotide ligation assay, which can be automated, and (2) the preferential homoduplex formation assay.
It is also possible to detect the mutations by PCR-based denaturating high-performance liquid chromatography, as described by Posteraro and colleagues.
Allele-specific primer polymerase chain reaction
Allele-specific primer (ASP)-PCR is especially useful to determine single nucleotide polymorphism in DNA samples, and this technique allows the identification of mutations without direct sequencing or digestion with restriction enzymes. This PCR format is based on the use of forward and reverse primers, which specifically anneal with the 2143G-mutated and 2142G-mutated sequences, respectively, of the 23S rRNA gene of H pylori . These primers are used with another primer pair designed upstream and downstream from the positions 2142 and 2143, respectively, to distinguish the wild-type A2142G and A2143G mutations from each other by amplicon sizes.
Using this method, Trespalacios and colleagues reported a 100% sensitivity and specificity for macrolide resistance in a series of 107 strains.
A modified version of the ASP-PCR was also proposed by Furuta and colleagues.
Primer mismatch polymerase chain reaction
This method uses 3′-end mismatch primers with the terminal nucleotide complementary only to the mutated nucleotide on the DNA template.
It was used to detect the A2142G, A2143G, and A2142C mutations in H pylori . This PCR was then extended to detect wild-type and 3 mutant genotypes. This method had a slightly better specificity than real-time PCR and a much better specificity than PCR–restriction length polymorphism (RFLP).
Invader assay for single nucleotide polymorphism genotyping
The Invader assay (or invasive signal amplification reaction assay) offers a simple diagnostic platform to detect single nucleotide changes with high specificity and sensitivity, originally used with unamplified genomic DNA. The Invader assay uses a structure-specific 5′ nuclease (or flap endonuclease) to cleave sequence-specific structures in each of 2 cascading reactions. The cleavage structure forms when 2 synthetic oligonucleotide probes hybridize in tandem to a target. One of the probes cycles on and off the target and is cut by the nuclease only when the appropriate structure forms. These cleaved probes then participate in a second Invader reaction involving a dye-labeled FRET probe. Cleavage of this FRET probe generates a signal, which can be readily analyzed by fluorescence microtiter plate readers. The 2 cascading reactions amplify the signal significantly; each original target molecule can lead to more than 10 6 cleaved signal probes in 1 hour. This signal amplification permits identification of single base changes directly, even from genomic DNA without previous target amplification. The sequences of the oligonucleotide components of the secondary reaction are independent of the target of interest and allow the development of universal secondary reaction components useful to identify any target.
This method was used by Furuta and colleagues on PCR amplicons (407 bp) of the 23S rRNA gene from gastric biopsies collected for the rapid urease test.
Non–polymerase chain reaction–based assays
Fluorescence in situ hybridization
The first report of FISH applied to the detection of H pylori resistance to clarithromycin was published by Trebesius and colleagues in 2000. It consists of an rRNA-based whole-cell in situ hybridization using a set of fluorescent-labeled oligonucleotide probes. Labeling of intact fluorescent bacteria is monitored by fluorescence microscopy. Using a 16S rRNA probe labeled with fluorochrome Cy3 (red) allows detection of H pylori , and simultaneously, a 23S rRNA probe labeled with fluorescein (green) detects the resistant mutants, which appear yellow by superposition of red and green.
This method proved to be sensitive and specific compared with standard methods of culture and antimicrobial susceptibility testing (AST).
Advantages
- •
This method does not need DNA extraction or amplification, therefore no apparati are needed, and it allows the bacteria-including coccoidal forms- to be visualized.
- •
It can be performed reliably on formalin-fixed material and therefore can be performed in a pathology laboratory.
- •
It allows a quick result.
- •
A test is commercially available, seaFAST H. pylori Combi Kit (SeaPro Theranostics International, Lelystad, The Netherlands).
Limitations are obtaining an observer-dependent result and sometimes it is difficult to read.
Peptide nucleic acid–fluorescence in situ hybridization
Peptide nucleic acid (PNA) probes using FISH can be designed to detect bacteria. PNA molecules are DNA mimics that have the negatively charged sugar-phosphate backbone replaced by an achiral, neutral polyamide backbone formed by a repetitive N -(2amninoethyl) glycine unit.
Specific hybridization between the PNA sequences and complementary nucleic acid sequences still occurs. The neutral PNA molecule is responsible for a higher thermal stability between PNA and DNA or RNA compared with the traditional DNA probes. Because of this situation, high-affinity PNA probes have sequences relatively smaller (13–18 nucleotides) than DNA sequences (>18 nucleotides) and are more resistant to nucleases and proteases than DNA molecules. They can be labeled by a fluorochrome dye and then detected by fluorescence microscopy or cytometry using the FISH method.
This method has been commonly used to detect H pylori in the environment, especially the aquatic environment, but it has also been applied to detect clarithromycin-resistant H pylori after culture and in histologic preparations from human gastric biopsies, with a sensitivity of 80% and specificity of 93.8%.
Furthermore, another variant of FISH named fluorescence in vivo hybridization has been developed using oligonucleotide variations comprising locked nucleic acids and 2′-O-methyl RNAs with 2 types of backbone linkages (phosphate or phosphorothioate). These probes hybridize at 37°C, with high sensitivity and specificity for H pylori , allowing visualization of these bacteria in biofilms.
Other methods
- •
Microelectronic chip assay: the assay performs multiple determinations, including identification of Helicobacter species ( H pylori vs H heilmannii ) and antibiotic resistance (macrolides and tetracycline), on the same microelectronic platform.
- •
Electrocatalytic detection of DNA sequences: the assay is based on electrocatalytic DNA detection assay, which reports DNA hybridization and resolves single-base changes in the target sequence. For example, an A2143C substitution significantly attenuates hybridization to an immobilized probe corresponding to the wild-type sequence. The single-base mismatch introduced by this mutation slows the kinetics of hybridization and permits discrimination of the 2 sequences when short hybridization times are used.
Correlation with Clinical Outcome
The main reason for discrepancies is essentially that genotypic methods are more accurate than culture plus antibiogram in detecting low numbers of resistant mutants in a population of susceptible bacteria, leading to several so-called heteroresistances (ie, a mixture of susceptible and resistant organisms) ( Table 2 ).
| Reference, Country | Treatment Used | N Patients | Genotypic Method | Eradication | Phenotypic Method | Eradication | ||
|---|---|---|---|---|---|---|---|---|
| Susceptibility (%) | Resistance (%) | Susceptibility (%) | Resistance (%) | |||||
| De Francesco et al, 2006 Italy | Clari Amox PPI and sequential | 146 | TaqMan real-time PCR | 94.5 (N = 91) | 46.1 (N = 13) 78.5 (N = 42) a | Etest | 92.4 (N = 119) | 55.5 (N = 27) |
| Liou et al, 2011 Taiwan | Clari Amox PPI | 303 | PCR sequencing | 90.8 | 28.6 | Agar dilution | 93.5 | 7.7 |
| Furuta et al, 2005 Japan | Clari Amox PPI | 139 | Invader assay on amplicon | 87.3 | 43.3 | Agar dilution | 87.3 | 43.3 |
| Lee et al, 2005 Korea | Clari Amox PPI | 114 | PCR-RFLP and sequencing | 79.8 (N = 91) | 0 (N = 23) | Etest | 79.8 (N = 91) | 0 (N = 23) |
Discrepancies were found between genotypic and phenotypic resistance. The concordance was 71.2% in 1 study and 80.6% in another, both using the TaqMan format of real-time PCR. Therefore, the question is: which method is the best predictor of the clinical outcome (ie, eradication of H pylori )?
According to De Francesco and colleagues, the best correlation is obtained with phenotypic results (Etest on H pylori strains) or with genotypic results not considering heteroresistance (TaqMan real-time PCR on paraffin-embedded gastric biopsies). Because the treatment used in this study was either standard clarithromycin-based triple therapy or sequential therapy, the latter may have a special impact on curing heteroresistant organisms in contrast to the former.
Liou and colleagues in Taiwan reported a result to the contrary in a subgroup of patients from a multicenter study (N = 303) comparing clarithromycin-based standard therapy with levofloxacin-amoxicillin-PPI. AST was performed on H pylori strains by agar dilution and a molecular method (PCR followed by sequencing of the 23S rRNA gene) on gastric biopsies. Contrary to what is established, these investigators found that the correlation between clarithromycin genotypic and phenotypic resistance was highest when the cutoff for resistance was set at MIC greater than 2 mg/L (κ correlation = 0.694). In the group of patients receiving clarithromycin, the eradication rates were 7.7% versus 93.5% for those harboring strains with or without 23S rRNA gene mutations (κ = 0.687) and 28.6% versus 90.8% for those harboring strains with MIC greater than 2 mg/L versus 2 mg/L or less, respectively (κ = 0.356).
The result may also depend on the type and the prevalence of the different mutations worldwide. The A2143G mutation may confer a higher risk of treatment failure.
Furuta and colleagues found the same proportion of H pylori eradication (ie, 87.3%) for susceptible versus 43.3% for resistant strains when resistance was determined phenotypically (agar dilution method) or genotypically (detection of the mutations by invasive signal amplification assay).
Lee and colleagues also found a perfect agreement between genotypic and phenotypic methods and furthermore a perfect correlation between clarithromycin resistance (or mutation) and lack of eradication and vice versa.
Related posts:
 Diagnosis of Helicobacter pylori Infection in the Proton Pump Inhibitor Era
Diagnosis of Helicobacter pylori Infection in the Proton Pump Inhibitor Era
 Gastric Cancer Risk in Patients with Helicobacter pylori Infection and Following Its Eradication
Gastric Cancer Risk in Patients with Helicobacter pylori Infection and Following Its Eradication
 Treatment Strategy for Gastric Mucosa-Associated Lymphoid Tissue Lymphoma
Treatment Strategy for Gastric Mucosa-Associated Lymphoid Tissue Lymphoma
 Helicobacter pylori
Helicobacter pylori
 When Is Endoscopic Follow-up Appropriate After Helicobacter pyloriEradication Therapy?
Screening to Identify and Eradicate Helicobacter pyloriInfection in Teenagers in Japan
When Is Endoscopic Follow-up Appropriate After Helicobacter pyloriEradication Therapy?
Screening to Identify and Eradicate Helicobacter pyloriInfection in Teenagers in Japan
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


