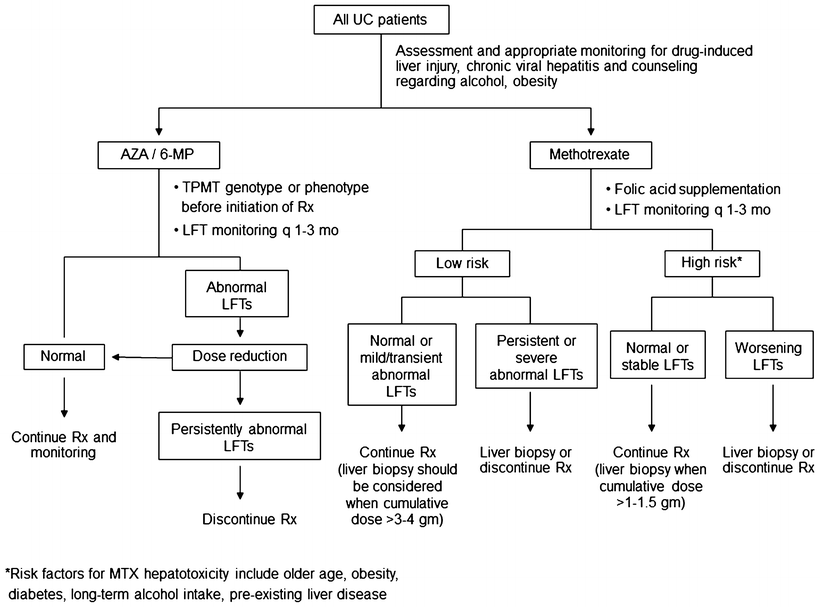
Fig. 41.1
Algorithm for the diagnosis of primary sclerosing cholangitis in patients with ulcerative colitis
Fig. 41.2
Typical endoscopic retrograde cholangiographic findings in primary sclerosing cholangitis
Both intra- and extrahepatic ducts are often involved (60–70 %), whereas localized intrahepatic duct (~25 %) or extrahepatic duct disease alone (<5 %) is less common [8, 48]. The cystic duct, pancreatic duct (PD), and gallbladder may be also involved. Severity of cholangiographic changes, scored by Amsterdam classification, has been noted to inversely correlate with transplant-free survival [51].
It should be noted that several conditions (i.e., ischemia, malignancy, chronic infection, and inflammation) can cause sclerosing and multifocal stricturing process of the biliary tract by nonimmune-mediated mechanism, and these may have cholangiographic features similar to those of PSC, the so-called secondary sclerosing cholangitis [8].
The findings on computer tomography (CT) and US are nonspecific. Thickening and/or saccular dilatations of the bile ducts and evidence of portal hypertension (i.e., varices, splenomegaly, and ascites) may be present. Contract enhancement of thickened bile duct wall is suggestive of an inflammatory process. Interestingly, abdominal lymphadenopathy, particularly in perihepatic and celiac axis groups, is often detected in PSC (66–100 %) and does not imply malignancy and should not exclude a patient from undergoing liver transplantation (LT) [52, 53].
In the presence of an abnormal cholangiogram, a liver biopsy is not required for the diagnosis of large-duct PSC. Periductal concentric (onionskin) fibrosis is a characteristic histopathologic feature. However, it is uncommonly encountered in a percutaneous biopsy specimen and may also be observed in secondary sclerosing cholangitis [8]. Importantly, liver biopsy may be essential to establish the diagnosis of small-duct PSC and PSC/autoimmune hepatitis (AIH) overlap as well as to exclude other causes of liver disease.
Natural History of PSC With and Without UC and Vice Versa
The clinical course of PSC is variable. The median time from diagnosis of PSC to death or LT has ranged from 9.6 to 21 years [1, 7, 26], and the overall survival is significantly decreased (approximately threefold) compared to the general population, even when asymptomatic at diagnosis [11, 16]. The clinical course is characterized by recurrent episodes of cholangitis, during which time the disease slowly progresses. Clinical features of pruritus and jaundice gradually develop overtime, and finally end-stage liver disease can appear [7]. Cholangiocarcinoma (CHCA) may complicate the course of PSC in 8–15 % of patients (annual incidence 0.6–1 %) [5, 7, 9]. Of interest is that the duration of PSC may not be a risk factor for CHCA, and, in fact, in approximately 50 % of patients with PSC plus CHCA, the malignancy is detected at the time of diagnosis or within the first year suggesting that superimposed CHCA may lead to the diagnosis of PSC [8, 54]. Compared to the general population, PSC patients are at higher risk for developing cancers (two- to tenfold for any cancers and 40–160-fold for colon cancer) [9, 11]. Patients with PSC are prone to develop complications of ESLD and portal hypertension (i.e., ascites, varices, encephalopathy). In some patients, esophageal varices may present early in the course of their liver disease, which is possibly explained by localized vascular damage in the portal triad from bile duct inflammation causing presinusoidal portal hypertension [5].
Serum bilirubin, albumin, and age at the diagnosis of PSC were independent prognostic factors in a time-dependent model [55]. Although the traditional Child-Pugh classification system is informative with regard to outcomes, the Mayo score model (includes age, bilirubin, AST, albumin, and history of variceal bleeding) may provide more reproducible and more accurate prognostic information without the need for liver biopsy, especially in patients with early disease [8, 56]. However, this model is not superior to the Child-Pugh system in predicting survival and related economic outcomes after LT [57]. The addition of cholangiographic findings in the model may provide some additional prognostic value [8, 48, 51].
The association between coexisting PSC and the disease extension and activity of UC remains inconclusive. UC patients with coexisting PSC tend to have higher incidence of pancolitis, backwash ileitis, and rectal sparing than UC patients without PSC [5]. However, patients with PSC-UC may have lower grade of colonic inflammation and more often run a quiescent course of colitis than UC patients without PSC [5, 58]. Colectomy with ileal pouch-anal anastomosis (IPAA) does not appear to alter the disease course of PSC [5].
Medical Therapy for PSC
A number of medical treatments that are targeted to alleviate inflammation and cholestasis have been investigated in PSC. However, unlike primary biliary cirrhosis (PBC), the efficacy of these therapies is somewhat limited. The uncertainty in the pathogenesis of PSC may present a barrier for the development of significant disease-modifying agents in PSC.
Ursodeoxycholic Acid (UDCA)
Ursodeoxycholic acid (UDCA) is a hydrophilic, tertiary bile acid which has been used for the treatment of a variety of chronic cholestatic conditions [59]. It has been shown to be an effective therapy in PBC [60]. After oral administration, UDCA is absorbed mainly in the small intestine, and then it has an enterohepatic circulation. At a daily dose of 13–15 mg/kg body weight/day, UDCA constitutes 40–50 % of total bile acid pool and results in a decrease in relative contribution of the more hepatotoxic endogenous hydrophobic bile acids [59]. Several mechanisms have been proposed by which UDCA may protect against cholestatic liver injury, and these include the choleretic effect by increasing bile flow, protection of injured cholangiocytes from toxic bile acids, stimulation of detoxification of hydrophobic bile acids, inhibition of hepatocyte apoptosis, and direct cytoprotective and immunomodulatory effects [29, 59].
The majority of the early studies of UDCA in PSC were small and/or uncontrolled. Many of these studies demonstrated liver function test improvement by using doses of 10–15 mg/kg body weight/day [8, 26, 61]. Lindor et al. conducted a randomized controlled trial (RCT) of UDCA 13–15 mg/kg/day, for 2–5 years, in 105 PSC patients. The results demonstrated improvement in LFTs but not symptoms and the time to treatment failure (defined by histologic progression by two stages, development of cirrhosis or esophageal varices, liver decompensation, LT, or death) [62]. On the basis that higher doses of UDCA may be required to provide sufficient delivery of UDCA to the bile pool and also enhance immunomodulatory effects in the setting of cholestasis and bile duct injury in PSC, several studies using higher dose of UDCA were conducted and published in the early 2000s. An RCT from Oxford using UDCA 20–25 mg/kg/day in 26 PSC patients found significant improvement in LFTs, histology, as well as cholangiographic features. However, no benefit in symptoms and survival was demonstrated [63]. Two studies comparing different doses of UDCA suggested that higher daily dose (25–30 mg/kg) was well tolerated and provided benefits, which included survival benefit in one study, compared to a lower dose (10–20 mg/kg) [64, 65].
Despite somewhat convincing data on benefits with higher doses, a large Scandinavian RCT evaluating UDCA at 17–23 mg/kg/day in 219 PSC patients for 5 years found no significant favorable effect on survival, symptoms, and prevention of CHCA although there was a nonsignificant trend toward improvement in LFTs and survival [66]. Recently, a multicenter RCT from the United States comparing high-dose UDCA (28–30 mg/kg/day) with placebo, in 150 PSC patients, was terminated prematurely at 6 years due to a higher incidence of adverse outcomes (i.e., death, LT, esophageal varices) in the UDCA group [67]. The likelihood of developing serious adverse outcomes was not predicted by biochemical response, but was predicted by advanced liver disease at presentation [67]. Therefore, currently there is no established role for UDCA in slowing the progression of PSC. Further, high-dose UDCA may be harmful in late-stage disease [8, 60].
Immunosuppressive Therapy
Unlike most of other immune-mediated diseases, treatment with corticosteroids and other ISAs has not demonstrated consistent benefits in PSC, and most evidence is derived from pilot studies. Corticosteroids demonstrated no benefit in PSC and were associated with significant worsening of osteoporosis [68–70]. Corticosteroids may be considered only in patients with PSC/AIH overlap and IgG4-associated cholangitis [8]. No controlled trial of azathioprine (AZA) has been reported as monotherapy to date. A combination of AZA, prednisolone, and UDCA (500–750 mg/day) for PSC was reported in a case series of 15 PSC patients. All patients had ALP improvement (seven patients had been previously treated with UDCA, but ALP improved only after prednisolone and AZA were added), and 60 % had histological improvement after 41 months [71]. Methotrexate (MTX) may minimally improve ALP levels, but does not impact clinical outcomes of PSC [72]. Addition of MTX to UDCA was associated with toxicity and without further improvement in LFTs [73]. Mycophenolate mofetil (MMF) was poorly tolerated (56 %) and did not demonstrate clinical benefit in PSC [74]. Further, combination of MMF and UDCA did not provide additional benefits [73]. Tacrolimus [75] and cyclosporin [76] had no significant effects on PSC disease outcomes and were poorly tolerated although they provided benefits in UC [76]. A pilot RCT in 10 PSC patients failed to demonstrate clinical efficacy of infliximab (5 mg/kg) on the course of liver disease [77].
Miscellaneous Treatment
Based on the observation that elevated hepatic copper levels are universally detected in patients with chronic cholestasis, D-penicillamine was evaluated in an RCT of 70 PSC patients. However, it was not associated with clinical benefit and has considerable toxicity, which led to treatment discontinuation in 21 % of patients [78]. Colchicine, an anti-fibrogenic agent, either alone or in combination with prednisone failed to show beneficial effects in two RCTs [70, 79]. Silymarin, a milk thistle extract, which potentially has several hepatoprotective properties, was evaluated in an open-label pilot study of 30 PSC patients for 1 year [80]. One-third of patients achieved substantial improvement in LFTs, but no significant change in Mayo risk score [80]. Several agents, such as nicotine, bezafibrate, pirfenidone, minocycline, and probiotics, have been preliminarily evaluated in PSC but without clear demonstrable benefits [8, 61, 81].
Medical Management for Complications of PSC
Cholangiocarcinoma (CHCA)
CHCA, a dreadful complication of PSC, develops in 8–15 % of patients [5, 7, 9]. Risk factors include the duration of UC, colonic dysplasia, variceal bleeding, proctocolectomy, alcohol consumption, and polymorphisms in the NKG2D gene [8, 82]. The diagnosis of CHCA in the setting of PSC is often challenging, particularly for the periductal infiltrative type. The presence of mass-like lesion or a long biliary stricture, particularly in the hilar area, on an imaging study strongly raises the possibility of CHCA. In PSC patients with clinical suspicion for CHCA, CA 19-9 at a cutoff of 129 U/mL has value in determining the likelihood for CHCA; positive predictive value was 57 % and negative predictive value 99 % [83]. However, caution must be exercised since CA 19-9 is undetectable in person with Lewis-negative blood type and can be elevated in other conditions (i.e., cholangitis, non-biliary cancers) [84]. A combination of cross-sectional liver imaging studies, tumor biomarkers, and cholangiography with tissue sampling is often required, and is recommended, to make the accurate diagnosis of CHCA (Fig. 41.3) [8, 54, 84].
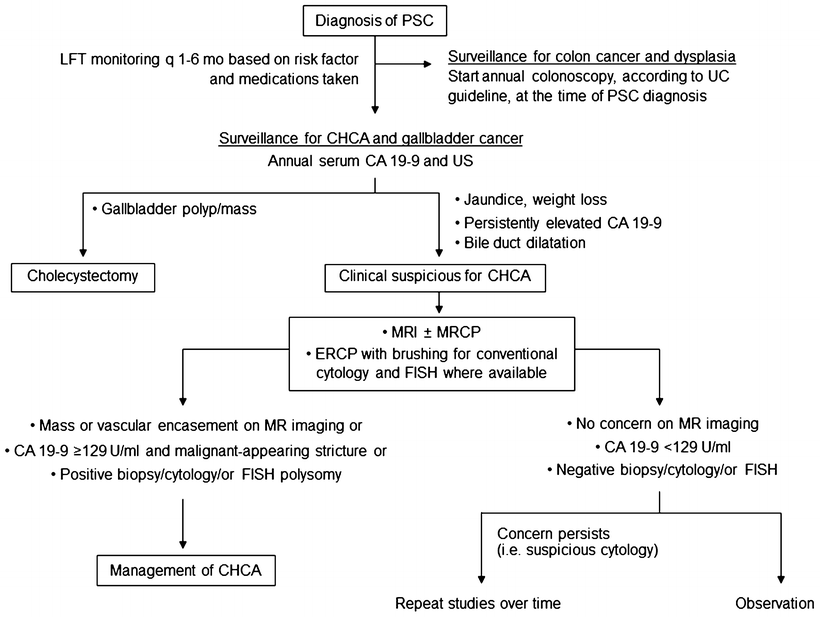
Fig. 41.3
Algorithm for the surveillance and the diagnosis of malignancy in patients with ulcerative colitis and primary sclerosing cholangitis
The prognosis of CHCA in PSC is dismal with a 3-year survival of less than 20 % even in surgically resected patients [8]. Recent data suggests that neoadjuvant therapy followed by LT in highly selected patients may result in a better outcome with a 5-year survival of ~70 % [85]. The benefit of other palliative modalities, such as external beam radiation, endoscopic ablative therapy, and systemic chemotherapy, has not been clearly demonstrated [8].
Colorectal Neoplasia
PSC has been shown to be an independent risk factor for the development of colorectal neoplasia in patients with UC (OR 4.79, 95%CI; 3.58–6.41) [86]. Colorectal neoplasia associated with PSC can be diagnosed at any time during the course of PSC, and it appears to occur predominantly in the right colon [87]. This risk appears to persist even after LT [5, 24]. Therefore, colonoscopy surveillance for colonic neoplasia is recommended to begin at the time of the diagnosis of PSC [8]. There are controversial data suggesting a benefit of UDCA in preventing the development of colorectal neoplasia [8, 60]. The current US guideline recommends against the use of UDCA as chemoprevention in patients with PSC-UC [8], while the European guideline suggests the use of UDCA in high-risk patients, such as those with strong family history of colorectal cancer, previous history of colorectal neoplasia, or long-standing extensive colitis [60].
Gallbladder Disease
Gallbladder abnormalities are commonly observed in patients with PSC, and these include gall stones (26 %), PSC involving the gallbladder (15 %), and gallbladder neoplasms (4–14 %) [8, 88]. Remarkably, 40–60 % of gallbladder polyps detected in patients with PSC are malignant [89, 90]. Therefore, surveillance by ultrasound should be done annually. In patients with a gallbladder mass lesion, cholecystectomy is recommended regardless of lesion size since the 1-cm rule may not reliably predict malignant potential of the gallbladder polyp in the setting of PSC [8, 91].
Bacterial Cholangitis
Patients with PSC are susceptible to repeated episodes of bacterial cholangitis, especially after biliary tract manipulation [92]. If cholangitis occurs without biliary intervention, the presence of stones, dominant strictures, or CHCA should be considered. Most common causative organisms are gram-negative enteric bacteria and enterococci [92]. The majority of patients respond to broad-spectrum intravenous antibiotic plus biliary drainage. Patients with recurrent bacterial cholangitis may benefit from long-term antibiotic prophylaxis [8].
Portal Hypertension and End-Stage Liver Disease (ELSD)
Management of portal hypertension and its complications in patients with PSC does not differ from other etiologies. The ultimate treatment for end-stage liver disease (ESLD) associated with PSC is LT with 5-year survival rates of approximately 85 % [8]. Resection of the extrahepatic biliary tree along with a Roux-en-Y choledochojejunostomy is widely accepted as a method of choice for biliary reconstruction in LT for PSC [60]. As in non-PSC, the Model for End-Stage Liver Disease (MELD) score is most widely utilized for organ allocation for PSC patients, although the presence of dominant strictures may affect MELD score by increasing bilirubin levels, and this may not necessarily mean advanced disease and liver failure. Other unique indications for LT in PSC patients include intractable pruritus, recurrent bacterial cholangitis, and CHCA [8]. Recurrence of PSC occurs in 20–25 % of the liver grafts after 5–10 years following LT [8, 93], but this is sometimes difficult to assess due to the similarities in biliary changes seen with ischemic and preservative injury, infections, and chronic rejection.
The activity of UC following LT is heterogeneous. Contrary to general wisdom, while on liver transplant-related immunosuppression, the majority of PSC-IBD patients experience a deterioration of their IBD following LT [94]. Further, the increased risk of developing colorectal neoplasia persists after LT [95]. The guideline for the management of UC exacerbation after LT has not been established, and the long-term effect of anti-TNF agents on liver graft is unknown.
Metabolic Bone Disease
Patients with long-standing IBD, and particularly with the prolonged use of corticosteroid therapy, frequently have decreased bone mass density (BMD) [96]. The presence of PSC, with or without cirrhosis, further negatively impacts BMD by several mechanisms, such as vitamin D malabsorption, altered bone turnover rate, and hypogonadism [97]. A recent study of 237 PSC patients with 10 years of follow-up reported that patients with PSC lost 1 % of their BMD per year. Osteoporosis was detected in 15 % of patients (24-fold higher rate than matched population) and risk factors included older age, low body mass index, and long duration of IBD [98]. The surveillance and management of osteoporosis in PSC does not substantially differ from other situations, and there is particular emphasis on calcium and vitamin D supplementation [8, 96]. Oral bisphosphonates may induce esophageal ulcerations which could precipitate variceal hemorrhage. Therefore, parenteral bisphosphonates may be a reasonable approach for patients with esophageal varices [8].
PSC Variants
Small-Duct PSC
Small-duct PSC, previously termed as pericholangitis, refers to a subgroup of patients who have biochemical and histological features compatible with PSC, but with normal cholangiography. Small-duct PSC represents approximately 6–11 % of patients with sclerosing cholangitis and often coexists with IBD (~80 %). It is a disease that is potentially progressive but is associated with a better long-term prognosis as compared with large-duct PSC (LT-free survival 13 years vs. 10 years, respectively; p < 0.0001) [99]. Cholangiocarcinoma does not seem to occur in patients with small-duct PSC. Approximately one-fourth of patients eventually progressed to large-duct PSC over a median of 7.4 years, and some patients progressed to end-stage liver disease requiring LT without developing large-duct disease [99]. Given a relatively small number of patients with small-duct PSC, the management is not well defined, and there is no controlled prospective study reported to date. In a longitudinal cohort of 42 patients from Mayo Clinic followed up to 25 years, UDCA 13–15 mg/kg/day improved liver biochemistries, but did not significantly delay disease progression [100].
PSC/Autoimmune Hepatitis (AIH) Overlap
PSC/AIH overlap is an ill-defined immune-mediated disorder, which is predominantly encountered in children and young adults [101]. A diagnosis of PSC/AIH overlap is made when both typical cholangiographic features of PSC and definitive diagnosis of AIH, based on modified AIH score, are present [101, 102]. The prevalence of PSC/AIH overlap in patients with PSC has varied from 7 to 14 % based on the revised AIH criteria, and majority of these patients (50–88 %) have underlying IBD [101]. The presentation of PSC/AIH overlap may be either simultaneous or sequential. Particularly in the setting of IBD, patients with PSC with an elevation of ALT should prompt a search for AIH. On the other hand, PSC should be considered in AIH patients with cholestasis, histological bile duct injury, and in those who show a poor response to therapy [101]. Patients with PSC/AIH overlap seem to benefit from UDCA and ISA, and survival is apparently better than in classical PSC, but with a poorer outcome than AIH [103, 105]. In a prospective Italian study (N = 7 PSC/AIH, 34 PSC), a combination of UDCA (15–20 mg/kg/day), prednisolone (0.5 mg/kg/day, then tapered to 10–15 mg/day), and azathioprine (50–75 mg) reported a good biochemical response (ALT, but not ALP) in patients with PSC/AIH overlap [104].
IgG4-Associated Cholangitis
IgG4-associated cholangitis (IAC) is a biliary tract disease of unclear pathogenesis and with cholangiographic features indistinguishable from PSC. It has been described in patients with autoimmune pancreatitis (AIP) as a part of a systemic autoimmune process associated with IgG4 [61, 105]. The clinical entity is characterized by pancreatic enlargement, elevated serum IgG4 levels, histologic evidence of lymphoplasmacytic infiltrate in the pancreas, and extrapancreatic manifestations, such as sclerosing cholangitis, sialadenitis, and retroperitoneal fibrosis [61, 105]. It is important to distinguish PSC from IAC, which is at times challenging due to the fact that pancreatic disease may not be evident (8 %) and IgG4-associated sclerosing colitis, mimicking IBD or UC itself, may be present [61, 105, 106]. IAC appears to be histologically distinct from PSC, and it usually has a dramatic response to corticosteroids in contrast to the refractory nature in PSC [5, 61]. All patients with possible PSC should be tested for serum IgG4 levels to exclude IAC [8]. Interestingly, up to one-quarter of patients with biopsy-proven PSC have an increased IgG4 periductal plasma cell infiltrate, and 9–22 % have elevated serum IgG4 levels [107, 108]. These findings raise the question of whether PSC and AIP/AIC represent different ends of the same disease spectrum or are distinct disease entities, although current evidence favors the latter [8]. Of note is that PSC patients with elevated IgG4 levels tend to have more severe disease severity and course [107, 108]. Thus, a trial of corticosteroids may be considered; however, proven benefit has yet to be demonstrated in RCT [61, 108].
Hepatobiliary Diseases Encountered in UC
Fatty Liver
Nonalcoholic fatty liver disease (NAFLD) is the most common hepatobiliary disease encountered in IBD, with reported prevalence range of 2–80 % (median 29.5 %) in published studies with over 100 subjects [109]. Thus, it may account for approximately 40 % of cases of LFT abnormalities in those with IBD [3]. As in the general population, the prevalence of NAFLD is likely increasing among IBD population as well [109]. Apart from obesity and metabolic syndrome, NAFLD can be caused by IBD-related factors, such as malnutrition, protein loss, and medications (i.e., corticosteroids, MTX, anti-TNFs) [109]. There is no specific guideline for the management of NAFLD in IBD patients; periodic monitoring and counseling to avoid excessive weight gain would be reasonable.
Portal Vein Thrombosis
Portal vein thrombosis (PVT) is rarely observed in IBD patients in a nonsurgical setting, but it is not uncommon in those with recent abdominal surgery [5]. Several factors associated with IBD, such as increased platelet counts, factor V, VII, and fibrinogen levels, state of active bowel inflammation, and infections, may contribute to the thrombotic complications [5]. Portal vein thrombosis has been detected by CT imaging in 25–45 % of UC patients who have undergone IPAA surgery and was more likely to be segmental, multiple, and occlusive [110–112]. Presentation of PVT following IPAA surgery includes abdominal pain, nausea/vomiting, prolonged ileus, and leukocytosis. Septic complications (i.e., liver abscesses, pelvic sepsis) subsequently occur in 0–50 % of patients [111, 112]. Anticoagulation and antibiotic treatments are generally associated with a good clinical response and resolution of PVT. Interestingly, postoperative PVT has been linked to an increased risk of subsequent pouchitis (46 % in patients with PVT and 15 % in those without after 36 months follow-up) [111].
Miscellaneous Conditions
Several other hepatobiliary diseases have been reported to be more prevalent in IBD patients than in the general population, and these include PBC, gallstones, hepatic amyloidosis, and granulomatous hepatitis [5] (Table 41.2). These conditions have been reported more often in CD than UC (with the exception of PBC), and the mechanisms are unclear. UC patients with PBC typically present with elevated ALP and positive AMA. Interestingly, unlike classical PBC, UC-associated PBC seems to occur at a younger age and more often in men [113]. Hepatic amyloidosis classically occurs in patients with long-standing active IBD and presents with elevated ALP and hepatomegaly (with hard consistency). Involvement of other organs, particularly gastrointestinal tract and kidneys, is commonly observed [114]. Granulomatous hepatitis can be linked to either IBD itself (CD) or IBD-related factors, such as medications, particularly sulfasalazine, and infections. It often presents with isolated elevation of ALP, although other features, such as fever and hepatomegaly, can also be encountered [5, 115]. The diagnosis of hepatic amyloidosis and granulomas generally requires a liver biopsy.
Hepatobiliary Diseases with Association to IBD Therapy (Tables 41.1 and 41.2)
Table 41.1
Hepatobiliary diseases associated with inflammatory bowel disease
UC | CD | |
|---|---|---|
Hepatobiliary disease with association to IBD or possibly shared pathogenesis with IBD | ||
• Primary sclerosing cholangitis (PSC) • Small-duct PSC • PSC/AIH overlap syndrome • IgG4-associated cholangitis | ++ ++ ++ ++ | + + + + |
Hepatobiliary disease encountered in IBD | ||
• Fatty liver disease • Portal vein thrombosis (PVT) • Presinusoidal portal hypertension • Gall stones • Hepatic amyloidosis • Granulomatous hepatitis • Primary biliary cirrhosis | ++ + ++ +/− +/− +/− ++ | ++ ++ +/− ++ ++ ++ + |
Hepatobiliary disease with association to IBD therapy | ||
• Drug-induced hepatotoxicity • Reactivation of hepatitis B • Hepatosplenic T-cell lymphoma | ++ ++ +/− | ++ ++ ++ |
Table 41.2
Differential diagnosis of hepatobiliary abnormalities associated with medications used in ulcerative colitis
Clinical features | Medications |
|---|---|
Acute hepatocellular injury | Sulfasalazine, 5-ASA, thiopurines, methotrexate |
Hypersensitivity reaction | Sulfasalazine, 5-ASA, thiopurines |
Autoimmune hepatitis features | Anti-TNF agents |
Cholestasis | Sulfasalazine, 5-ASA, thiopurines, cyclosporin |
Cirrhosis | Methotrexate |
Steatohepatitis | Methotrexate |
Nodular regenerative hyperplasia (NRH) | Thiopurines |
Non-cirrhotic portal hypertension | Thiopurines |
Sinusoidal obstruction syndrome (SOS) | Thiopurines |
Peliosis hepatis | Thiopurines |
Pancreatitis | Thiopurines |
Reactivation of hepatitis B | Anti-TNF agents, corticosteroids, thiopurines, methotrexate, cyclosporin |
Hepatosplenic T-cell lymphoma | Anti-TNF agents (typically in combination with thiopurines) |
Drug-Induced Liver Injury (DILI)
5-Aminosalicylic Acid (ASA) Compounds
Sulfasalazine-induced DILI in IBD is relatively uncommon, with three severe cases per million prescriptions reported in the United Kingdom [116]. Acute hepatocellular damage may develop alone or, less commonly, as a part of generalized hypersensitivity reaction, which is characterized by fever, malaise, lymphadenopathy, and hepatomegaly. It has been suggested that DILI, particularly a hypersensitivity reaction, is related to the sulfapyridine moiety; however, recent data reported similar incidence of overall DILI with sulfasalazine (sulfapyridine and 5-ASA) and mesalamine (5-ASA alone), suggesting that DILI is more likely from 5-ASA rather than the sulfa moiety [116, 117]. Cross-hypersensitivity reaction with mesalamine after a prior hypersensitivity reaction to sulfasalazine has been reported [118]. Hepatotoxicity can occur from as early as 6 days to 1 year after initiation of therapy. Other forms of DILI include granulomatous hepatitis, acute liver failure, chronic hepatitis, and cholestasis [5, 115].
Thiopurines
Treatment of IBD with azathioprine (AZA), 6-mercaptopurine (6-MP), or 6-thioguanine (6-TG) can be associated with various forms of DILI, mainly acute hepatocellular injury, idiosyncratic cholestatic reaction, and hepatic vascular endothelial injury [3, 115, 119]. Recent data reported a prevalence of thiopurine-induced acute DILI in IBD patients to be 3.4–7.1 %, with an annual incidence of 1.4–2.6 % [3, 119]. Liver function test abnormalities are usually reversible and often occur soon after the initiation of treatment [115, 119–121]. In addition, hypersensitivity reaction and cholestatic hepatitis have also been reported [115]. Hepatotoxicity from AZA/6-MP is related to its metabolite 6-methylmercaptopurine ribonucleotide (6-MMP); however, the sensitivity and specificity of 6-MMP levels for DILI were poor [122]. In an effort to avoid the potential adverse events from AZA/6-MP, assessment of thiopurine methyltransferase (TPMT) genotype or phenotype before initiation of treatment is suggested [120, 123]. Individuals with low TPMT activity are at risk for myelotoxicity via higher levels of 6-TG, whereas individuals with high TPMT activity have lower 6-TG levels and can result in a suboptimal treatment response, as well as hepatotoxicity from higher levels of 6-MMP [115, 120, 123]. With close monitoring, allopurinol co-therapy with low-dose AZA/6-MP may help to avoid poor response and decrease risk of hepatotoxicity, especially in patients with very high TPMT activity [124]. In addition, split-dosing regimen of thiopurines, by decreasing 6-MMP levels, has been proposed to reduce the risk of DILI [122].
Most cases of idiosyncratic cholestatic reaction associated with AZA/6-MP occur within 2–5 months of treatment and with a male preponderance. Jaundice may not immediately resolve despite drug withdrawal [119].
Apart from direct hepatic injury, thiopurines can be associated with hepatic vascular endothelial lesions and their consequence, and these included nodular regenerative hyperplasia (NRH) and non-cirrhotic portal hypertension, sinusoidal obstruction syndrome (SOS or formerly called venoocclusive disease), and peliosis hepatis [5, 115]. Nodular regenerative hyperplasia has been reported to occur from 1.3 to 18 % of IBD patients who receive 6-TG, and it may develop as soon as few months or many years after therapy [125, 126]. The development of NRH seems to be dose related, as it is rarely seen with low-dose 6-TG (<20 mg/day) [115]. Male gender and small bowel resection of >50 cm appear to be additional risk factors [125]. Majority of patients are asymptomatic with mild abnormality in LFTs. The definite diagnosis of NRH is based on a liver biopsy, while MRI has a sensitivity of 77 % and a specificity of 72 % [126]. The natural course of NRH-associated with thiopurines is usually indolent and potentially reversible. However, adverse outcomes, such as portal hypertension, varices, and hepatocellular carcinoma, have also been reported [115].
Methotrexate
Long-term use of MTX has been associated with liver fibrosis and cirrhosis in patients with psoriasis and rheumatoid arthritis, and often LFTs are normal. Risk factors for DILI from MTX include high cumulative dose, older age, alcohol, obesity, diabetes, and preexisting liver disease [5]. Since there is poor correlation between ALT and histologic changes, surveillance with liver biopsy is recommended to monitor for MTX hepatotoxicity, traditionally after a cumulative total dose of 1.5 g. Despite limited data, the incidence of MTX hepatotoxicity in IBD patients appears to be lower than in those with psoriasis and rheumatoid arthritis. In a series of 20 IBD patients receiving a cumulative MTX dose of 1.5–5.4 g, only one patient developed hepatic fibrosis on biopsy [127]. Concordantly, in a recent series of 87 IBD patients, MTX was commonly associated with LFT abnormalities (24 %), but these frequently normalized while still on therapy, and in only 5 % was drug discontinuation necessary. Among patients with LFT abnormalities, 44 % had underlying risk factors for DILI, and liver biopsy rarely showed substantive abnormalities [128]. Therefore, liver biopsy may not routinely be necessary for IBD patients without risk factor(s). Close monitoring with LFTs every 1–3 months is recommended, and liver biopsy should be performed if the majority of ALT values over 1-year period are elevated or if serum albumin is decreased (Fig. 41.4) [5, 123, 129]. Supplementation with folic acid may help to reduce hepatic adverse effects associated with MTX [130].