Chapter 98C Liver transplantation in children and segmental transplantation
Historical Overview
Thomas Starzl attempted the first human liver transplantation in 1963 in a 3-year-old child with biliary atresia. Unfortunately, the child died in the operating room from uncontrollable hemorrhage (Starzl, 1992), but subsequently, in 1967, Starzl successfully performed liver transplantation in eight children. All survived the operation, and half survived more than 1 year (Starzl et al, 1968).
In 2007, there were 5887 adult liver transplantations and 602 pediatric transplantations (Organ Procurement and Transplantation Network/Scientific Registry of Transplant Recipients [OPTN/SRTR], 2008). Currently, approximately 15,500 adults and 540 children are awaiting liver transplantation (United Network for Organ Sharing [UNOS], 2009). Potential recipients who are infants, toddlers, or young children have narrow constraints with regard to graft size. Historically, the lack of size-matched organ availability, coupled with the disproportionate number of adults on the waiting list, has significantly disadvantaged children awaiting transplantation. Compared with adults who have liver disease, children have much less time to await transplantation; an infant with decompensated liver disease from cirrhosis typically cannot survive on a waiting list for several years (McDiarmid, 2002). Furthermore, waiting-list mortality for children younger than 1 year continues to far exceed that of other children and adults (OPTN/SRTR, 2008), in part because of the lack of size-matched organs.
Progressive improvements in surgical technique have evolved such that now the left lateral section and the right trisegment graft each can be used, meaning that a single liver potentially can be divided and transplanted into two recipients. With this innovative technique, the split-liver procedure, important progress in the effort to expand donors available to children has been accomplished without disadvantaging adult recipients. Except in unusual circumstances, the use of a reduced-size liver allograft in which the remnant is discarded is theoretically rarely justified. In 2007, partial or split-liver transplantations accounted for approximately 4% of all deceased-donor liver transplantations (OPTN/SRTR, 2008). The use of reduced-size liver grafts declined from 23% to 16% of pediatric liver transplantations over the last decade, but during this same time, split-liver allografts increased from 9% to 16% (Anderson et al, 2008).
Favorable outcomes transplanting reduced-size and split livers led to the development of techniques for using liver segments from living donors for pediatric grafts. With increasing experience, donations from living donor livers are no longer confined to left lateral sections or left hepatic lobes but have expanded to include full right hepatic lobe grafts, which contributes to the adult organ pool as well (Hong et al, 2008). Today, living-related and unrelated living-donor liver transplantation (LDLT) accounts for approximately 10% of all pediatric liver transplantations and 4% of adult liver transplants (OPTN/SRTR, 2008), and it has been one of the most exciting and challenging advances in liver transplantation.
Indications
Box 98C.1 lists common indications for pediatric liver transplantation. Biliary atresia is a progressive, inflammatory, fibrosing cholangiopathy of unclear pathogenesis (see Chapter 40). It occurs in only 1 in 8000 to 18,000 live births; and although it is rare, it remains the most common cause of infant death owing to hepatic disease and accounts for approximately 50% of all pediatric liver transplantations (Bassett & Murray, 2008; Bezerra, 2005; Perlmutter & Shepherd, 2002). Most children with this disease lack an extrahepatic biliary tree, resulting in impaired bile flow, conjugated hyperbilirubinemia, acholic stools, and hepatomegaly. The hepatic parenchyma becomes congested, and progressive damage leads to secondary biliary cirrhosis. Although portoenterostomy, also known as the Kasai procedure (Kasai & Suzuki, 1959), may yield some clinical improvement and is the first-line treatment (Ohi, 2000; Ryckman et al, 1998), approximately 70% to 80% of children with biliary atresia eventually require liver transplantation (Karrer et al, 1996; Lowell et al, 1996).
Box 98C.1 Indications for Pediatric Liver Transplantation
Other causes of cholestasis in children include idiopathic neonatal hepatitis; infection by both viral and bacterial pathogens, including toxoplasmosis and syphilis; progressive familial intrahepatic cholestasis (Byler disease); metabolic and genetic diseases; familial arteriohepatic dysplasia (Alagille syndrome); choledochal cyst; and ischemia-reperfusion injury. Cholestasis may progress to cirrhosis and has also been associated with parenteral nutrition in children with short bowel syndrome (Baker et al, 1998).
Pediatric liver transplantation is also indicated in cases of noncholestatic liver failure secondary to infectious, metabolic, genetic, and neoplastic etiologies. Although less frequent in children than in adults, postnecrotic liver cirrhosis is the indication for transplantation in approximately 10% of pediatric patients and is most commonly the result of viral hepatitis or idiopathic cryptogenic cirrhosis (Malatack et al, 1983).
Metabolic liver diseases are genetic disorders that lead to the production of aberrant transport proteins or enzymes and altered metabolic pathways. These inborn errors of metabolism, such as α1-antitrypsin (AAT) deficiency or Wilson disease, may cause direct injury to the liver and result in liver failure, with or without injury to other organs; it may also lead to abnormal liver metabolism, including urea cycle disorders or oxalosis, that can lead to injury of other organs. Altogether, such diseases account for approximately 10% of all pediatric liver transplantations (Hansen & Horslen, 2008).
AAT deficiency is a genetic disorder that leads to defective production of the serine protease α1-antitrypsin (see Chapter 70A), an enzyme produced in the liver to protect the lungs from neutrophil elastase. The abnormal protein accumulates in the liver, apparently causing liver damage, although the mechanism of injury remains unclear. Synthetic AAT has some utility in the treatment of AAT deficiency–related lung disease, but it has no role in the treatment of the associated liver disease (Köhnlein & Welte, 2008; Silverman & Sandhaus, 2009).
Wilson disease is an inborn error of metabolism characterized by defective copper excretion and the accumulation of toxic amounts of copper in the liver, basal ganglia, kidney, and cornea, which may lead to the characteristic Kayser-Fleischer rings (see Chapter 70A). The defect is located on chromosome 13, is inherited in an autosomal recessive fashion, and affects 1 in 50,000 births. Initial symptoms are nonspecific and include lethargy, anorexia, vague abdominal pain, and weight loss. Some patients present with asymptomatic hepatomegaly, and others present with fulminant hepatic failure (FHF) (Riordan & Williams, 2001).
CN syndrome is caused by mutations of the gene that codes uridine diphosphate glucuronosyltransferase-1, resulting in unconjugated hyperbilirubinemia. Untreated neonates develop a condition of severe neuronal damage called kernicterus. Homozygous familial hypercholesterolemia (HFH) is a rare disease caused by mutations in the gene that codes the low-density lipoprotein (LDL) receptor. The receptor may be defective or completely absent, and affected individuals have dramatically elevated plasma cholesterol levels, leading to accelerated atherosclerosis, childhood coronary artery disease, and premature death from myocardial infarction (Hansen & Horslen, 2008). Liver transplantation corrects these conditions by providing the required cellular machinery to synthesize the correct gene product or metabolite (Florman & Shneider, 2001; Meyburg & Hoffmann, 2005). Occasionally, auxiliary hepatic transplantation has been performed in these circumstances (Bismuth et al, 1996; Sze et al, 2009; Van Hoek et al, 1999). In AAT deficiency, the potential for development of hepatocellular carcinoma (HCC) precludes auxiliary liver transplantation and mandates total hepatectomy.
Cystic fibrosis (CF) is an autosomal recessive multiorgan disorder that is the most common lethal inherited disease affecting the white population. As management of these patients continues to improve, CF liver disease (CFLD) is increasingly recognized as a significant cause of morbidity and mortality. Approximately 5% to 10% of CF patients will progress to cirrhosis, with the most frequently recognized complications attributable to portal hypertension with an associated nutritional and pulmonary decline. A significant number of patients with CFLD may be helped with an isolated liver transplantation or a concomitant liver and lung transplantation; however, the debate is ongoing in the literature regarding the timing of liver transplantation in these patients (Colombo et al, 2006; Fridell et al, 2003; Nash et al, 2008).
Although more common in adult patients, chronic Budd-Chiari syndrome with severe hepatic congestion and focal areas of liver fibrosis or cirrhosis may be an indication for liver transplantation in older children (see Chapter 77). Pretransplantation evaluation should be performed to recognize predisposing factors or underlying diseases such as myeloproliferative disorders, primary hepatic protein deficiencies—of proteins C and S, antithrombin III, and activated protein C resistance—or secondary protein deficiencies, such as increased intestinal protein loss in inflammatory bowel disease (Slakey et al, 2001).
FHF (see Chapters 73 and 97C) is associated with a mortality rate greater than 70%. It is essential to transfer children with FHF to a transplant center immediately for management and urgent evaluation and listing for liver transplantation. Because cerebral edema develops rapidly, careful monitoring and intensive supportive care are required. Mild elevation of intracranial pressure (ICP) may be managed acutely by hyperventilation and elevation of the patient’s head, although maintenance likely requires strict sodium and osmolar and intravascular volume control. Placement of an ICP monitor should be considered, but regardless, attention to maintenance of cerebral perfusion pressure (CPP) is paramount. Transplantation in patients with signs of cerebral edema is urgent, as edema may lead to irreversible brain damage or death (Tanaka et al, 1994). Poor prognostic factors for liver transplantation in children with FHF include age younger than 10 years, liver disease other than viral hepatitis, grade 2 or 3 hepatic encephalopathy, coagulopathy (prothrombin time >30 seconds), and increasing jaundice (bilirubin >9 mg/dL) (Uemoto et al, 2000).
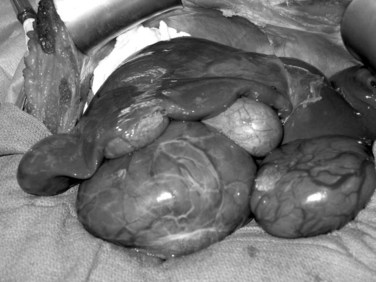
Liver tumors account for less than 3% of all indications for pediatric liver transplantation (Austin et al, 2006). Hepatoblastoma, HCC, and fibrolamellar HCC represent the most frequent tumors (see Chapter 82). In contrast to adults, the predisposing factors for malignant liver tumors in children are not viral hepatitis or alcoholism but are more often metabolic disorders, such as AAT deficiency, tyrosinemia, or glycogen storage disease. In patients without cirrhosis, hepatic resection is the treatment of choice. Liver transplantation should be considered in the case of unresectable tumors confined to the liver (Chen et al, 2006; Otte et al, 2004). In patients with advanced cirrhosis and HCC meeting Milan criteria, transplantation is indicated (Austin et al, 2006; Beaunoyer et al, 2007; Perilongo et al, 2004). Rarely, giant arteriovenous malformations and benign liver tumors—when they replace the whole liver, or when they have the potential for malignancy (e.g., hepatic adenomatosis or multiple adenomas in glycogen storage disease [Fig. 98C.1])—may be indications for total hepatectomy and liver transplant (Malatack et al, 1987; Wellen et al, 2009; see Chapter 79A).
Evaluation of the Potential Recipient
A multidisciplinary evaluation of a child with decompensated liver disease should be completed with the involvement of surgeons, hepatologists, nurses, anesthesiologists, psychologists, and social workers. Early referral to a transplant center allows maximum time to develop a management strategy and to optimize pretransplantation clinical status. The timing of liver transplantation is crucial, because late referral of patients with significant hepatic complications and malnutrition results in a poorer outcome. The expected waiting time on the transplant list may vary based on the geographic region, and this should be taken into account. In infants younger than 2 years who weigh less than 10 kg, it is often difficult to maintain metabolic and nutritional support. Ideally, children should be considered for liver transplantation before the complications of malnutrition, such as weight loss and growth failure, occur (Kimura et al, 2004; McDiarmid et al, 2004). With chronic liver disease, prolonged clotting times, intractable ascites, recurrent variceal bleeding secondary to portal hypertension, and recurrent cholangitis with severe cholestasis are indications for liver transplantation (Hendrickson et al, 2004).
The prognosis after liver transplantation for chronic liver disease is influenced by preoperative comorbidities. The advent of the Pediatric End-Stage Liver Disease (PELD) and Model for End-Stage Liver Disease (MELD) scores (Malinchoc et al, 2000; Wiesner et al, 2001) has made it possible to better predict mortality and progression of liver disease while awaiting transplantation (Desai et al, 2004; Barshes, et al, 2006). The pertinent factors in the PELD score are the international normalized ratio (INR), total bilirubin, serum albumin, age younger than 1 year, and weight or length less than 2 standard deviations (SDs) from the mean for age and gender. The PELD score is used for patients younger than 12 years, and the scores range from a negative value (−10) to 50. Adolescents aged 13 to 18 years are allocated organs based on the MELD score, similar to adults. Box 98C.2 presents the formulas for calculating the MELD and PELD scores.
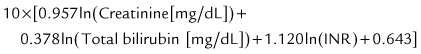
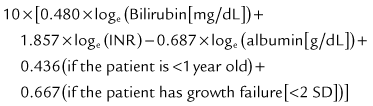
Comparing the periods before and after the introduction of the MELD and PELD scores, a similar percentage of children are being transplanted, but mortality rates have decreased (Freeman et al, 2004; Yao et al, 2004). Small size and young age are no longer contraindications, and many transplantations for biliary atresia are now done in infants who weigh less than 10 kg (Colombani et al, 1996; Sokal et al, 1990). Whereas posttransplantation mortality has improved, waiting-list mortality for infants younger than 1 year remains high, unfortunately (OPTN/SRTR, 2008). Consideration should be given to referring these young and small infants to pediatric specialty centers that have expertise with these challenging cases. Contraindications to pediatric hepatic transplantation are listed in Box 98C.3.
Recipient Hepatectomy
Before beginning the transplantation, a Broviac central venous catheter is placed for intraoperative and postoperative fluid resuscitation, monitoring, and postoperative laboratory draws, the same as an arterial catheter. Pressure points are well padded, and convective heating blankets are used. The abdomen is entered through a curved transverse subcostal incision. Meticulous hemostasis must be maintained throughout the procedure, because ongoing bleeding may lead to dilutional coagulopathy and fibrinolysis. Abdominal wall venous collaterals must be carefully controlled as they are encountered, and adhesions to the liver are carefully dissected to allow access to the portal region (Fig. 98C.2).
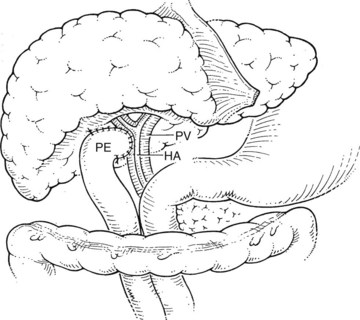
For orthotopic whole-liver transplantation, the small retrohepatic caval segment can be removed with the native liver, or the vena cava can be preserved and the donor liver anastomosed in a piggyback fashion to the confluence of the recipient’s right, middle, and left hepatic veins. With a segmental graft, the recipient inferior vena cava must be preserved. A vascular clamp is placed across the hepatic veins so that they can be joined to form a common opening to sew to the donor suprahepatic cava or, in case of segmental grafts, the left hepatic vein (Fig. 98C.3). Complete hemostasis of the retroperitoneum should be accomplished before removing the donor liver from cold storage.
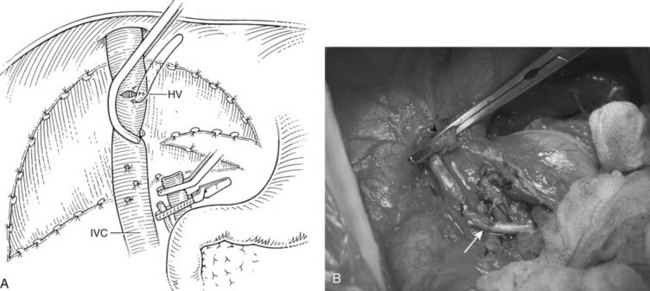
Graft Procurement and Engraftment
The techniques used for recovering whole, deceased-donor livers for transplantation from children are similar to the techniques used in adult-adult transplantation. Special attention should be paid to donor serum sodium levels and to dynamic liver function tests. High sodium levels (>170 mmol/L) may be associated with severe reperfusion injury and poor graft function (Totsuka et al, 1999).
The arterial anastomosis is performed with sutures of fine (7-0 or 8-0) monofilament material using microsurgical loupes. A microscope may be useful in anastomosing arteries with a diameter smaller than 1 to 2 mm. The anastomosis is performed most commonly in an end-to-end fashion directly to the native hepatic artery. If the native artery is not of adequate size, or if there is an intimal dissection, an extension allograft using saphenous vein or donor iliac artery from the infrarenal or supraceliac aorta may be used (Yamaoka, 1996).
Reduced-Size Segmental Liver Transplantation and Split-Liver Transplantation
Reduced-Size Segmental Liver Transplantation
The use of reduced-size grafts for pediatric recipients is one of the most important advances in liver transplantation. The disproportionate shortage of grafts for very young children, compared with older children, motivated surgeons to develop innovative operative techniques to overcome the restrictions imposed by donor and recipient size mismatch. With these techniques, livers can be reduced to a functional unit of appropriate size for the recipient. The optimal ratio between the required volume of liver and the actual “tailored” hepatic mass is between 1 : 1 and 1 : 2. In this way, livers from donors 10 or 20 times the size of the recipient can be used (Broelsch et al, 1988).
The liver comprises eight anatomically defined segments, each with its own arterial and portal blood inflow and separate biliary drainage (Strasberg, 1997; see Chapter 1B). Similarly, segmental hepatic venous outflow is independent, but major liver veins run along sectional borderlines, receiving blood from several adjacent segments (Couinaud, 1954). For purposes of transplantation, the liver can be divided into several functional grafts. The most commonly used reduced-size graft is the left lateral section graft, composed of segments II and III (Fig. 98C.4; Botero & Strasberg, 1998). To preserve a single donor portal vein, the liver is transected just to the right of the falciform ligament, and branches of the ascending (umbilical) portion of the left portal vein to segment IV are carefully suture ligated. The left hepatic vein is used for hepatic venous drainage. It is anastomosed directly to the recipient’s vena cava, which must be preserved during hepatectomy. The full left graft (segments I, II, III, and IV or segments II, III, and IV) is used in small adults or older children who require a graft slightly larger than the left lateral section.
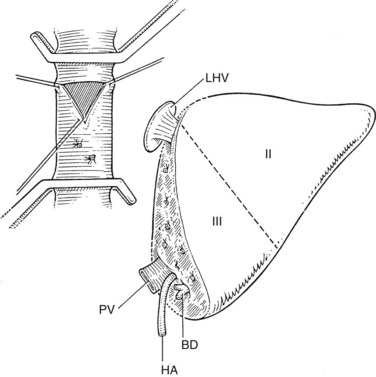
Bismuth and Houssin (1984) first reported success with transplantation of a left hepatic allograft; in the same year, Broelsch and colleagues (1984) reported the successful grafting of the left lateral section. Since then, most pediatric transplant centers have adopted segmental transplant techniques using reduced-size livers. The results after these segmental grafts are comparable to the results achieved with whole-organ orthotopic liver transplants (Hong et al, 2008
Related posts:
 Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
 Portal hypertension in children
Portal hypertension in children
 Chemotherapy and radiotherapy for pancreatic and periampullary cancer: Adjuvant, neoadjuvant, and palliative
Chemotherapy and radiotherapy for pancreatic and periampullary cancer: Adjuvant, neoadjuvant, and palliative
 Bile duct exploration and biliary-enteric anastomosis
Bile duct exploration and biliary-enteric anastomosis
 Interventional endoscopy: Technical aspects
Interventional endoscopy: Technical aspects
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


