Chapter 5 Liver regeneration
Mechanisms and clinical relevance
Liver Regeneration
In contrast to other solid organs, the human liver has the unique ability to regenerate after toxic injury, chronic inflammation, and surgical resection. The terms regeneration, hyperplasia, and hypertrophy are used synonymously in literature, but hyperplasia is the most precise term from a cellular standpoint. The damaged hepatic lobes do not grow back in the same way a lizard’s tail regrows, but rather a hyperplastic response is seen, defined as increasing cell numbers in the remnant liver, leading to its hypertrophic appearance and enlargement of the liver (Lory et al, 1994). This process is highly regulated and involves multiple cell types, extrahepatic signals, complex molecular pathways, and cellular interactions. A delicate balance is achieved in the initiation of regeneration and exertion of an intracellular growth response to restore lost liver mass, while maintaining the normal metabolic function necessary for survival. The inability to maintain this process may lead to poor liver function and ultimately liver failure after surgery.
Clinical Relevance of Liver Regeneration
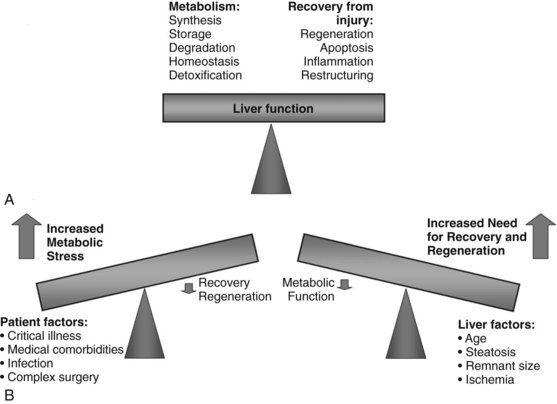
Today, liver surgery is routinely and safely accomplished for malignant and benign disease. This substantial progress in both resection and transplantation relies on the ability of the liver to regain most of its functional mass within 2 weeks (Marcos et al, 2000a; Humar et al, 2004; Kamel et al, 2003; Pomfret et al, 2003). Factors that limit the achievement of curative tumor resection and small-for-size (SFS) transplantation are the high morbidity and mortality rates associated with insufficient volume of the liver remnant or graft (Fig. 5.1).
As hepatobiliary and transplant surgeons continue to expand the magnitude and complexity of liver resection and explore beyond the boundaries of transplantation, understanding liver regeneration becomes increasingly relevant. Many tumors once considered unresectable are now amenable to complete resection through induction chemotherapy and innovative treatment strategies to increase liver remnant volume (see Chapters 86 and 88) (Clavien et al, 2007). Portal vein embolization (PVE) or ligation causes atrophy of the ipsilateral hemiliver and hypertrophy of the contralateral side and appears to be particularly valuable in patients who have underlying liver disease (see Chapter 93A). Because of the increased success of liver surgery and improved anesthetic techniques, more patients with underlying liver disease are now considered suitable candidates for liver resection.
There remains, however, a subset of patients who cannot tolerate significant liver resection, often because of the inability of the liver to regenerate appropriately. Patients with underlying cirrhosis have increased morbidity and mortality (Das et al, 2001; Nagasue et al, 1987), and patients with metabolic syndrome, morbid obesity with concomitant steatosis, or nonalcoholic steatohepatitis (NASH) have increased risk of liver failure after liver surgery (Belghiti et al, 2000; McCormack et al, 2007) as do elderly patients with restricted hepatic reserve and decreased regenerative capacity (McCormack et al, 2007). In colorectal malignant disease, about 50% of patients will develop liver metastases in the course of their disease. With substantially improved chemotherapy for colorectal cancer over the last decade, the resectability rate has gradually increased to 20% to 30% (Khatri et al, 2005). As a consequence, more patients are having liver surgery after neoadjuvant or induction chemotherapy, which also has considerable side effects on liver regeneration.
Regeneration is also crucial in liver transplantation (see Chapter 97A). In deceased-donor transplantation, hepatocyte loss occurs in the form of ischemia/reperfusion (I/R) injury owing to the necessary preservation period from procurement to implantation and damage that may have occurred in the donor. Due to the scarcity of organs, more marginal organs are accepted for transplantation; this has increased the need for regeneration in the environment of I/R injury and use of immunosuppressive drugs. One of the landmark advances in liver transplantation is the ability to use partial liver grafts obtained from either a deceased donor or a living donor. In the latter setting, success of the procedure relies on hepatic regeneration in both donor and recipient. The minimal amount of functional liver necessary for successful transplantation is a major concern. Because of the need for regeneration, as well as recovery from concomitant I/R injury, a larger liver mass is needed for transplantation than might be required after liver resection (Tanaka & Yamada, 2005).
Basic Characteristics of Liver Regeneration
Models of Liver Regeneration
In the past 50 years, much has been learned about what initiates liver regeneration, how it is sustained, and what inhibits it. Liver regeneration is most clearly shown in the experimental model that was pioneered in 1931 by Higgins and Anderson. In this model, a simple two-thirds partial hepatectomy (PHx) was performed, without damage to the lobes left behind. This led to enlargement of the residual lobes, which made up for the mass of the removed lobes in 5 to 7 days. Other well-known models of liver regeneration are associated with extensive tissue injury and inflammation and include the use of hepatic toxins—such as ethanol (EtOH) (Kaplowitz, 2002), carbon tetrachloride (CCl4) (Reynolds, 1963), and galactosamine (GalN) (Dabeva & Shafritz, 1993)—bile duct ligation (Kountouras et al, 1984) or portal vein ligation (Bilodeau et al, 1999), and I/R injury (Jaeschke, 1998). Newer models include transgenic albumin promoter urokinase-type plasminogen activator (u-PA) fusion constructs (Heckel et al, 1990) and PHx in Zebrafish (Kan et al, 2009). In each model, different toxic agents injure specific liver cell subpopulations; PHx is therefore the preferred in vivo model to study the regenerative response. It has also been demonstrated that regenerative signaling observed in a rat liver transplant model of I/R injury is similar to that observed after PHx (Debonera et al, 2001).
General Features of Liver Regeneration
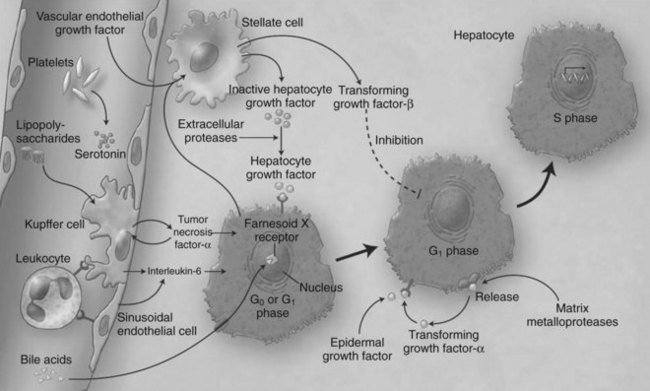
Liver regeneration after surgical resection is carried out by proliferation of existing mature cellular populations. These include hepatocytes, biliary and fenestrated endothelial cells, Kupffer cells, and cells of Ito (stellate cells; Fig. 5.2) (Gressner, 1995). The kinetics of cell proliferation and the growth factors produced by proliferating hepatocytes suggest that hepatocytes provide the mitogenic stimuli that lead to proliferation of the other cells; the degree of hepatocyte proliferation is directly proportional to the degree of injury (Bucher & Swaffield, 1964).
Immediately after liver resection, the rate of DNA synthesis in hepatocytes begins to increase, as they exit the resting state of the cell cycle (G0) and enter G1, traverse to DNA synthesis (S phase), and ultimately undergo mitosis (M phase). The induction of DNA synthesis occurs later in the nonparenchymal cells, at about 48 hours for Kupffer and biliary epithelial cells and at about 96 hours for endothelial cells (Grisham, 1969; Widmann & Fahimi, 1975). The first peak of DNA synthesis occurs at 40 hours after resection in rodents and at 7 to 10 days in primates. In small animals, the regenerative response returns the liver to the preresection mass in 1 week to 10 days. Clinical studies suggest that significant regeneration in humans is evident within 2 weeks after resection and is nearly complete at 3 months (Nagasue et al, 1987; Yamanaka, 1993). Studies in living donors, however, suggest that many donors do not reach 100% of starting liver volume by 3 months after donating their right lobe, and some are even lacking at one year (Pomfret et al, 2003; Nadalin et al, 2004; Ibrahim et al, 2005).
On a cellular level, hepatocyte proliferation starts in the periportal areas of the lobules (Rabes et al, 1976) and proceeds to the pericentral areas by 36 to 48 hours. Liver histology at day 3 to 4 after PHx is characterized by clumps of small hepatocytes surrounding capillaries, which change into true hepatic sinusoids. The hepatic matrix composition also changes from high laminin content to primarily containing fibronectin and collagen types I and IV. After a 70% hepatectomy, restoration of the original number of hepatocytes theoretically requires 1.66 proliferative cycles per residual hepatocyte. In fact, most of the hepatocytes in the residual lobes (95% in young and 75% in very old rats) participate in one or two proliferative events (Stocker & Heine, 1971). However, hepatocytes have an almost unlimited capacity to regenerate, and transplantation of several hundred healthy hepatocytes can repopulate a whole, damaged liver in a calculated minimum of 69 doublings (Rhim et al, 1994).
Liver Stem Cells
Endogenous Hepatic Progenitor Cells
In this situation a population of cells known as oval cells proliferates to replace the hepatic parenchyma (Fausto & Campbell, 2003). In humans, hepatic progenitor cells (HPCs) participate in repopulation of the liver after acute massive necrosis and have also been identified in chronic liver disease (Roskams et al, 1998). The oval cells are thought to derive from biliary epithelial cells, and human HPCs seem to originate from the canals of Hering (Theise et al, 1999) and play an important role in acetaminophen-induced injury (Kofman et al, 2005). Recent data suggest that in addition to mature hepatocytes and HPCs, a third compartment of cells can participate in liver regeneration (Alison et al, 2009; Duncan et al, 2009). Several studies in rodents and humans have demonstrated that transplanted bone marrow stem cells can enter the liver from the circulation and give rise to hepatocytes and cholangiocytes (Petersen et al, 1999; Theise et al, 2000a; Alison et al, 2000). These stem cells usually make little contribution to regeneration, but after fusing with metabolically defective hepatocytes, they can be reprogrammed to contribute in a major way to restoring liver function or to stimulating liver regeneration via paracrine effects (Lagasse et al, 2000; Wang et al, 2003a; Kallis et al, 2007).
Exogenous Mobilized Progenitor Cells
Mesenchymal stem cells (MSCs) isolated from human umbilical cords (Yan et al, 2009), adipose tissue (Banas et al, 2008), and bone marrow (Kuo et al, 2008; Abdel Aziz et al, 2007) have been shown to promote liver repair in cases of acute liver damage (e.g., carbon tetrachloride injections). The major fraction of mobilized progenitor stem cells expresses the chemokine receptor CXCR4. At the same time, the mRNA level of its ligand, SDF-1, was increased in the damaged liver tissue. These results provide a clue that CXCR4–SDF-1 interaction may be important for the mobilization of progenitor stem cells from bone marrow to the damaged liver (Khurana & Mukhopadhyay, 2007). In the damaged liver, these MSCs could generate cells that function as normal hepatocytes, but a second mechanism by which MSCs could promote liver repair is secretion of soluble factors. For instance, infusion of human MSC-conditioned medium into rats with acute liver damage improved liver function after 24 hours (Parekkadan et al, 2007; van Poll et al, 2008) with a 90% decrease in apoptotic hepatocytes and a threefold increase in the number of proliferating hepatocytes. Very recently, human liver-derived stem cells were shown to accelerate hepatic regeneration after hepatectomy by shuttling mRNA from microvesicles into the regenerating hepatocytes (Herrera et al, 2009).
Induction of Proliferation: Priming and Cell-Cycle Progression
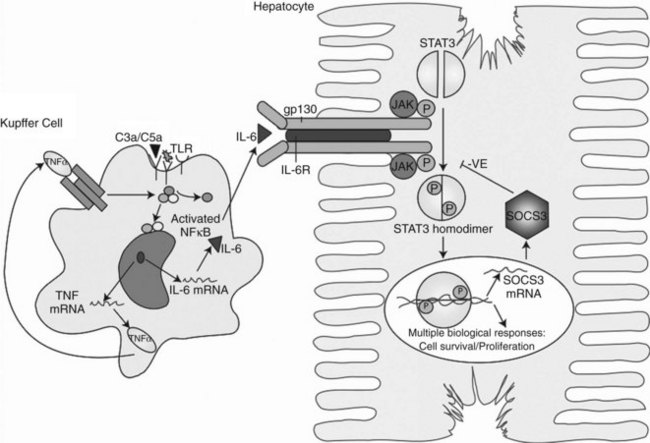
Within minutes after PHx, specific immediate early genes are activated in remnant hepatocytes (Haber et al, 1993). These genes include protooncogenes, which play an important role in normal cell-cycle progression—such as in JUN, FOS, MYC, and KRAS (Thompson et al, 1986; Morello et al, 1990; Taub, 2004)—and the transcriptional factors nuclear factor (NF) κB, signal transducer and activator of transcription 3 (STAT3), activator protein-1 (AP-1), and CCAAT enhancer–binding protein β (CEBPβ) (Cressman et al, 1995; FitzGerald et al, 1995). Historically, the onset of liver regeneration has been attributed to a flow-dependent response, by which increased relative flow after PHx resulted in hepatocyte proliferation and hyperplasia (Starzl et al, 1977). Experiments in parasymbiotic rats demonstrated the existence of humoral factors in the induction of liver growth after PHx (Moolten & Bucher, 1967). Interleukin (IL)-6 and tumor necrosis factor (TNF)-α have since been identified as the earliest factors triggering activation of several transcription factors during regeneration (Fig. 5.3) (Cressman et al, 1996; Yamada et al, 1997). In IL-6–deficient or TNF-α receptor–deficient mice, liver regeneration after hepatectomy is delayed (Streetz et al, 2000) but not completely abolished (Michalopoulos et al, 1984; Fujita et al, 2001). Therefore, other blood-derived mitogens, such as hepatocyte growth factor (HGF), were identified as a putative hepatic growth factor during liver regeneration (Fausto, 2000). Hepatocytes in normal liver will not respond to mitogenic signals without a set of “priming” events that switch them into a responsive state. This has been described by Webber and colleagues (1994), who identified priming factors, involved in initiating and triggering hepatic response to injury, and concomitant growth factors and their receptors, which allow competent hepatocytes to progress through the cell cycle. Priming is accomplished by the release of preformed cytokines that subsequently activate transcription factor complexes and allow the cell to exit G0 into G1 in the cell cycle. This group includes TNF-α and IL-6 (Aldeguer et al, 2002). Growth factors include the potent hepatocyte mitogens HGF, transforming growth factor (TGF)-α, and heparin-binding epidermal growth factor (HB-EGF). This process is further controlled by comitogens—such as insulin, glucagon, steroid and thyroid hormones, and epinephrine—that facilitate activity of the mitogens and by downregulation of growth factor inhibitors, such as activin A and TGF-β.
So far, few factors have been identified as possibly responsible for the release of these priming cytokines and growth factors in the onset of liver regeneration. The first is endotoxin lipopoly-saccharide (LPS), produced in the gut by gram-negative bacteria. Circulating LPS appears to be a strong signal for Kupffer cells to start the cascade that results in hepatocyte replication. Rats treated with antibiotics and germ-free rodents have a delayed peak of DNA replication after PHx, confirming the importance of LPS (Cornell et al, 1990). Another major finding is the demonstration that cytokine activation and DNA replication are severely impaired in mice lacking the complement components C3a and C5a (Strey et al, 2003). In particular, mice lacking both C3a and C5a have impaired production of TNF-α and IL-6 after PHx and poor activation of NFκB and STAT3. In the initiation of growth factors, urokinase-type plasminogen activator (uPA) appears to play an important role; uPA and its downstream effector, plasminogen, increase within 1 to 5 minutes after PHx and rapidly cleave the HGF precursor, pro-HGF. Blocking uPA delays the appearance of HGF and thereby delays liver regeneration, whereas blocking plasminogen-activator inhibitor (PAI) accelerates the release of HGF and accelerates liver regeneration (Currier et al, 2003).
More recently, new important factors in the initiation of liver regeneration have been identified. Extracellular adenosine triphosphate (ATP) has emerged as a rapid-acting signaling molecule that leads to quick and transient activation of JNK signaling after PHx, induction of immediate early genes c-FOS and c-JUN, and activator protein 1 (AP-1) DNA-binding activity (Thevananther et al, 2004; Gonzales et al, 2009). Also recently, an important role for blood platelets in I/R injury and after partial liver resections has been demonstrated. Thrombopenic animals exhibited reduced hepatocyte proliferation 48 hours after the initial ischemic insult, with diminished expression of TNF-α and IL-6 as early as 4 hours after reperfusion (Nocito et al, 2007). In a model of partial hepatectomy, the effect was shown to be mediated by serotonin, because mice lacking platelet serotonin displayed a lack of liver regeneration (Lesurtel et al, 2006). About 95% of all serotonin found in blood is stored in platelets and in vitro, and serotonin is a potent mitogen that stimulates hepatocyte mitosis (Balasubramanian & Paulose, 1998). The platelet effect on liver regeneration after PHx was mediated through the 5-HT2A and 2B receptor subtypes on hepatocytes and could be restored by injection with the serotonin precursor 5-HTP. These results suggest that a molecular action of serotonin is involved in the induction of hepatoctye proliferation after a major loss of hepatic tissue and that it may involve the release of growth factors, such as IL-6 (Clavien, 2008).
Intracellular Pathways and New Profiling Techniques
The importance of liver regeneration in sustaining life is characterized by the plethora of pathways involved and the fact that liver regeneration is usually only blunted or delayed in experimental models that lack a “critical” pathway. There are nonetheless several well-described patterns in liver regeneration. The immediate early genes that are activated in regenerating liver were first identified by Mohn and colleagues (1990) and Haber and colleagues (1993), who described induction patterns of more than 70 genes activated in the first week after two-thirds PHx in a rat model, genes normally not expressed in the quiescent liver. The development of cDNA microarray technologies set the stage for studying genome-wide expression and expanded this list to about 180 genes (Su et al, 2002; Arai et al, 2003).
A mouse cDNA array has also been used to study the crucial role of IL-6 in liver regeneration in IL-6–deficient mice after hepatectomy, showing a 36% reduction in the early gene expression compared with wild-type mice. In the absence of IL-6, activation of genes such as u-PA receptor (PLAUR) and SERPINE1, crucial for HGF activation and the MAPK pathway, were remarkably delayed (Li et al, 2002). IL-6 activates the JAK/STAT3 and MAPK signaling pathways via the gp130/IL-6R complex. This leads to activation of an array of immediate and delayed early genes required for normal liver-specific metabolic functions, repair, and protection from injury (Taub, 2003; Zimmers et al, 2003; Wuestefeld et al, 2003). STAT3 is crucial for cells to progress from the G1 to S phase and for activating the MYC gene required for cell-cycle progression. Other intracellular signaling pathways that involve the receptor tyrosine kinases p38 MAPK, pERKs, and c-jun N-terminal kinase (JNK) are also rapidly activated.
Progression through the cell cycle is regulated by cyclins and cyclin-dependent kinases (CDKs). Various combinations bind to form kinase complexes that are active at distinct points within the cell cycle and tightly controlled by several mechanisms, including binding by CDK-inhibitory proteins, such as p21. Feedback signals to this process are provided by the suppressor of cytokine signaling-3 (SOCS3) and TGF-β, also regulated by IL-6. Other studies confirmed the importance of NFκB (Arai et al, 2003; Fukuhara et al, 2003; Togo et al, 2004) and showed immediate upregulation of apoptotic genes (FAS and caspases) in livers that failed owing to excessive resection (Fig. 5.4) (Morita et al, 2002). The cytokine-induced form of nitric oxide synthase (iNOS) seems to plays an important role in scavenging oxygen radicals and offering protection from apoptosis caused by a proinflammatory response mediated through IL-6 and TNF-α (Rai et al, 1998).
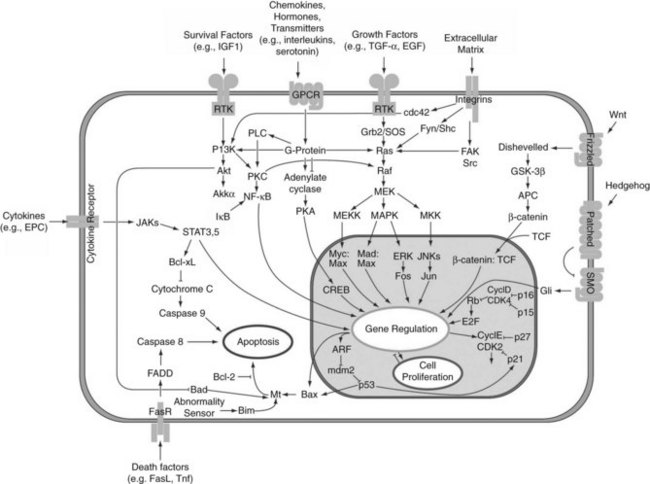
FIGURE 5.4 Intracellular pathways of liver regeneration. Multiple intracellular signaling pathways are rapidly activated. Progression through the cell cycle is regulated by cyclins and cyclin-dependent kinases (CDKs) tightly controlled by several mechanisms, including binding by CDK-inhibitory proteins, such as p21. (From http://en.wikipedia.org/wiki/File:Signal_transduction_pathways.png.)
The HGF/c-Met pathway is important for sustaining DNA synthesis after injury and activates various downstream pathways that involve PI3K, ERK/MAPK, and AKT1 (Huh et al, 2004). This pathway cross talks with the wnt/β-catenin signaling pathway, which has come to the forefront in liver biology over the last several years (Monga, 2009). Increased levels of HGF results in β-catenin dissociation along with nuclear translocation (Monga, 2002) and upregulation of downstream targets of this pathway, such as cyclin D1, MYC, PLAUR, matrix metalloproteinases (MMPs), and epidermal growth factor receptor (Michalopoulos & DeFrances, 1997). Vascular endothelial growth factor (VEGF) interacts with endothelial cells in the liver to increase HGF production from nonparenchymal cells (LeCouter et al, 2003).
In the transplant setting, microarray analysis of SFS rat liver grafts showed upregulation of vasoconstrictive and adhesion molecule genes at early time points postreperfusion, with later increases in genes associated with inflammation and cell death and downregulation of genes related to energy metabolism (Man et al, 2003), which was recently confirmed in the situation of clinical deceased-donor and living-donor liver transplantation (de Jonge et al, 2009).
Remodeling of the Liver
Remodeling of the newly regenerated liver tissue begins with the repopulation and maturation of nonparenchymal cells, such as endothelial cells, stellate cells, and biliary epithelial cells. Newly formed hepatocytes form clusters into which replicating endothelial cells invade to form new sinusoids. To restore normal architecture, stellate cells, located between endothelial cells and hepatocytes, synthesize extracellular matrix proteins and TGF-β1, which can regulate the production of hepatic extracellular matrix (see Chapter 6). VEGF, angiopoietins 1 and 2, TGF-α, fibroblast growth factors (FGFs) 1 and 2, and HGF all are likely involved in the angiogenic process. Angiostatin, an inhibitor of angiogenesis, causes delayed and suppressed liver regeneration in mice (Drixler et al, 2002; Vogten et al, 2004). Remodeling of the extracellular matrix is associated with the activation of the urokinase-plasminogen pathway and the membrane-bound matrix metalloproteinase (MMP) pathway. MMP-9 is one of the most active proteins during liver remodeling and regeneration (Kim et al, 2000). The MMPs—in combination with HGF, EGF, and TGF-β1—act to remodel the extracellular matrix, changing the levels of several extracellular matrix proteins such as collagen, fibronectin, laminin, and entactin. The maturation and thickening of the extracellular matrix seems to have an inhibitory effect on proliferating hepatocytes, potentially signaling the end of rapid regeneration (Kim et al, 1998).
Maintaining Liver Function During Regeneration
After volume loss, hepatocytes must adapt rapidly and seek a compromise between maintenance of continued differentiated function and cellular replication to permit survival. After toxic injury, resection, or transplantation, the balance is dramatically shifted to the crucial tasks of recovery and regeneration at the expense of normal hepatic metabolism. The success of restoring lost liver mass, repairing tissue injury, and resolving inflammation determines the ability of the liver to support normal metabolic function and determines its ability to recover (Fig. 5.5) (Strey et al, 2004). Several of the expressed immediate early genes encode enzymes and proteins involved in regulating gluconeogenesis, a critical process after PHx to compensate for the lost glycogen content and to produce sufficient glucose for the whole organism (Haber et al, 1995; Rosa et al, 1992). Rapid and increased expression of genes involved in glucose homeostasis is seen after PHx in animal models. Most notably, these include PCK, glucose-6 phosphatase (G6Pase), and IGFBP1, controlled at the level of transcription by insulin (downregulation), glucagon and cAMP (upregulation), and glucocorticoids (upregulation) (Mohn et al, 1990; Diamond et al, 1993). Livers lacking IGFBP1 display abnormal liver regeneration (Leu et al, 2003).
Decreased expression of genes that oppose gluconeogenesis also occurs rapidly posthepatectomy. Insulin itself can be a potent growth factor mediated through the insulin receptor, and insulin and glucagon have long been established as important “gut-derived” growth factors (Starzl et al, 1975). Liver-specific transcription factors—hepatocyte nuclear factors (HNFs)—play an important role in determining the level of glucose production, fatty acid metabolism, and liver-specific secreted proteins. CAAT/enhancer binding protein (C/EBP) α regulates expression of genes involved in hepatic glucose and lipid homeostasis, has antiproliferative proprieties, and is downregulated during liver regeneration after hepatectomy (Mischoulon et al, 1992; Costa et al, 2003).
During early regeneration, the liver accumulates fat. Neither the mechanisms responsible for nor the functional significance of this transient steatosis has been determined. Suppression of hepatocellular fat accumulation is associated with impaired hepatocellular proliferation after PHx, indicating that hepatocellular fat accumulation is specifically regulated during, and may be essential for, normal liver regeneration (Shteyer et al, 2004). Some data show that decreasing lipid peroxidation levels by vitamin treatment after PHx produces an attenuation of cellular apoptosis and a marked increase in the proliferation process, suggesting that the modulation of lipid peroxidation has a role in the liver regeneration process (Ronco et al, 2002). Hepatocytes in the periportal regions that divide and replicate after PHx require mitochondrial fatty acid β-oxidation. Peroxisome proliferator-activated receptor α, PPARα, may be a crucial modulator, controlling energy flux important for repair of liver damage and regeneration (Anderson et al, 2002). Expression of many other liver-specific genes, such as IGFBP1 and G6Pase, is regulated in the basal state by hepatic nuclear factor-1 (HNF1A). The transcriptional activity of HNF1A is upregulated during liver regeneration by the binding of HNF1A to the growth-induced transcription factors STAT3 and AP-1 (Leu et al, 2001). Together, growth-induced and tissue-specific transcription factors enable the liver to maintain metabolic function, despite the loss of two thirds of its functional mass.
In a microarray analysis of gene expression profiles after living-donor liver transplantation, it was recently demonstrated that C/EBPα was immediately downregulated, as was as HNF4α and PPARα (de Jonge et al, 2009). The immediate, marked downregulation of genes associated with pathways of lipid, fatty acid, and carbohydrate metabolism demonstrate how a liver segment needs to balance regenerating efforts with metabolic needs. It is this balance that limits the size of liver segment that can be transplanted or the size of remnant liver after donation or resection.
Termination of Proliferation
The size of the liver is highly regulated and is controlled by the functional needs of the organism. The most well-known antiproliferative factors within the liver are TGF-β and related family members such as activin (Derynck & Zhang, 2003). TGF-β is produced mainly by hepatic stellate cells, but in the early phase, it forms inhibitory complexes with SNoN and SKI proteins (Macías-Silva et al, 2002), rendering hepatocytes initially resistant to TGF-β (Koniaris et al, 2003). Later on, TGF-β acts through a heteromeric receptor complex, which subsequently phosphorylates proteins of the SMAD family, particularly SMAD2 and SMAD3 (Lonn et al, 2009). The SMAD2/SMAD3-SMAD4 complex translocates into the nucleus and activates target genes that negatively regulate the cell cycle (Nakao et al, 1997). Reactive oxygen species (ROS) enhance synthesis and activation of TGF-β (Koli et al, 2008), which may account for the reduced regeneration after ischemia and reperfusion. Interacting with the TGF-β/SMAD signaling could restore regeneration in a model of SFS liver grafts (Zhong et al, 2010).
Similarly, activin A blocks hepatocyte mitogenesis and shows diminished signaling during liver regeneration when its cellular-receptor level is reduced; its receptor level is restored once liver regeneration is terminated (Date et al, 2000). The level of activin-receptor mRNA expression was shown to be an important determinant in the magnitude of regeneration in portal vein ligation and PHx (Takamura et al, 2005a, 2005b). Suppressors of cytokine signaling (SOCS) are important negative regulators of cytokine signaling that prevent the tyrosine phosphorylation of STAT proteins. It has been shown that IL-6 signaling in the liver causes rapid upregulation of SOCS3, which correlates with the subsequent downregulation of phosphorylated STAT3, thereby terminating the IL-6 signal (Campbell et al, 2001).
Recent work strongly implicates the detection of blood bile acid levels by nuclear receptors as a regulator of liver growth (Huang et al, 2006). In addition, mammalian genes comparable to those in the Drosophila Hippo kinase-signaling cascade, which regulates wing mass during development, can also control hepatocyte proliferation (Dong et al, 2007). Overexpression of YES1-associated protein (YAP), the mammalian counterpart to yorkie (yki)—the last gene in the Drosophila Hippo kinase cascade—in a transgenic mouse model leads to massive liver hyperplasia, reaching 25% of body weight. YYA1P1 transcriptionally activates cell-cycle proteins, such as Ki-67 and c-Myc, and also activates inhibitors of apoptosis, and its phosphorylation by way of activation of the Hippo pathway blocks its ability to shuttle to the nucleus (Reddy & Irvine, 2008). It is therefore possible that the Hippo kinase pathway has a decisive role in determining overall liver size (Duncan et al, 2009).
Liver Atrophy
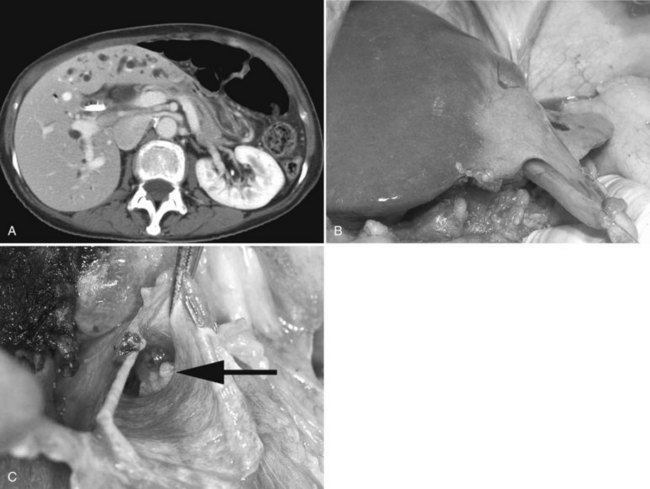
Classically, atrophy is triggered by an obstruction of portal venous blood flow or is the result of chronic obstruction of the bile duct. When atrophy occurs unilaterally, the opposite lobe of the liver responds with a hypertrophic response, which may result in a rotation of the liver around the hilar axis. The liver can become grossly distorted with marked changes in anatomic landmarks, most commonly seen accompanied by a rotation of the portal triad structures (Fig. 5.6). In recent years, the anatomic changes accompanying atrophy led surgeons to utilize preoperative embolization of the portal vein in areas involved with malignancy, hoping to induce hypertrophy in areas of the contralateral liver to increase the potential for resection and to reduce postoperative morbidity (see Chapter 93A).
Mechanisms of Liver Atrophy
The cause of liver cell death in atrophy is generally divided between necrosis and apoptosis. The distinction is important, because necrosis is a nonregulated traumatic disruption of a cell that occurs when it encounters overwhelming injury, whereas apoptosis is an inducible, highly orchestrated cascade of physiologic events. Necrotic cells lose membrane integrity, leak lysosomal enzymes, and induce a substantial inflammatory response. Apoptosis is energy dependent and allows cells to shrink and die without inducing inflammation. Apoptotic cells are characterized by plasma membrane blebbing, chromosomal condensation, and nuclear DNA fragmentation (Steller, 1995).
Portal Vein–Induced Hepatic Atrophy
The most common clinical scenario of hepatic atrophy is portal vein–induced atrophy, wherein occlusion of the portal vein leads to ischemia in areas of the liver; atrophy in this setting is a result of liver cell death secondary to necrosis and apoptosis (Ikeda et al, 1995; Shibayama et al, 1991). Ischemic necrosis of centrilobular areas of the liver predominates in the first 3 days of cell death. Areas peripheral to the necrotic liver cells predominantly undergo apoptotic cell death, and apoptosis persists long after necrosis subsides. Oxygen levels and mitochondrial function help determine which cells will undergo necrosis or apoptosis (Nishimura et al, 1998). Models of portal vein ischemia in rats have confirmed a caspase-dependent apoptosis and have indicated that Kupffer cells are involved in generating reactive oxygen substrates and other acute-phase reactants, culminating in mitochondrial dysfunction and apoptosis (Kohli et al, 1999).
Biliary-Induced Hepatic Atrophy
The molecular mechanisms involved in biliary obstruction leading to hepatic atrophy are much more centered on apoptosis, with little or no involvement of acute necrosis. Cholestasis results in the accumulation of toxic bile salts, which induce apoptosis through the Fas-mediated pathway. In this case, TNF-α and Fas ligand bind to the Fas death receptors, leading to a cascade of intracellular events, including cytochrome-c release from mitochondria and activation of apoptosis-mediating caspases. In Fas-deficient mice, bile duct ligation resulted in impaired apoptosis and less injury and fibrosis compared with wild-type mice (Canbay et al, 2002; Gujral et al, 2004; Miyoshi et al, 1999). More recent data, however, suggest a nonischemic model of necrosis/oncosis as the predominant process leading to cell death after common bile duct ligation, with cell swelling and without apoptotic caspase 3 activation (Fickert et al, 2005).
Clinical Causes of Atrophy
Hilar cholangiocarcinoma is a frequent cause of biliary atrophy, induced by occlusion of a major bile duct. Biliary occlusion, often accompanied by portal vein compromise, leads to atrophy of the liver approximately 20% of the time with this disease and has significant surgical implications. Also, postcholecystectomy bile duct strictures lead to hepatic atrophy 10% to 15% of the time (Hadjis & Blumgart, 1987; Pottakkat et al, 2009). Choledocholithiasis and hepatolithiasis are infrequent causes of atrophy, as are benign tumors such as papillomas, cystadenomas, and granular cell tumors (Blumgart et al, 1984; Blumgart & Kelley, 1984). Strictures caused by parasitic biliary infections, such as by Clonorchis sinensis and Ascaris lumbricoides, also have been known to cause biliary obstruction and associated atrophy of the liver. Occlusion of the hepatic artery alone would not induce atrophy.
Compensatory Regeneration Triggered by Atrophy
In the embolized portion of the liver, Kupffer cells release TNF-α and induce hepatocyte swelling and hyperplasia in the contralateral liver. Additionally, increased portal flow in the nonembolized lobe induces proliferation and activates several cytoplasmic growth-promoting signal-transduction pathways involved in hepatic regeneration, including JUN and the mitogen-activated protein kinase pathways. Kupffer cells in the contralateral liver begin to produce TNF-α as well, stimulated either by increased portal flow or through the direct effect of circulating growth factors (Kim et al, 2001). Nonparenchymal cells in the injured portion of the liver produce HGF. Messenger RNA levels of HGF in the ischemic portion of liver were noted to be 10 to 20 times that of normal, with most upregulation occurring in Kupffer cells (Hamanoue et al, 1992).
Factors Influencing Liver Regeneration
Patient-Related Factors
Age
Liver age is a significant factor in hepatocellular regeneration. Older livers do not regenerate as quickly as younger livers and show delayed regeneration after acute injury and impaired function after liver transplantation (Burroughs et al, 2006; Feng et al, 2006). Rodent models have shown reduced and delayed thymidine kinase uptake in older animals after PHx, and a striking difference is seen in the magnitude of DNA synthesis and timing of hepatocyte replication between young and old livers (Taguchi et al, 2001). Treatment of old mice with growth hormone could restore hepatocyte proliferation (Krupczak-Hollis et al, 2003; Jin et al, 2009). Aging also represses activation of the c-Myc promoter in old livers after hepatectomy and in tissue culture models(Timchenko, 2009; Iakova et al, 2003).
On a clinical level, little is known regarding differential gene expression in old versus young livers, but results from the United Network for Organ Sharing (UNOS) database show reduced resistance in old donor livers to prolonged cold ischemia times (Cassuto et al, 2008), possibly as a result of high mitochondrial susceptibility to oxidative stress (Lenaz et al, 2000). In living-donor transplantation, some studies have shown that increasing donor age significantly affects posttransplant function (Abt et al, 2004; Iida et al, 2009) and rate of regeneration in the donor (Yokoi et al, 2005).
Biliary Obstruction
Cholestasis results in the accumulation of toxic bile salts, which induce apoptosis through the Fas-mediated pathway. In this pathway, TNF-α and the Fas ligand bind to the Fas death receptors, leading to cytochrome-c release from mitochondria and activation of apoptosis-mediating caspases. In Fas-deficient mice, bile duct ligation resulted in impaired apoptosis and less injury and fibrosis compared with wild-type mice (Canbay et al, 2002; Gujral et al, 2004; Miyoshi et al, 1999). Other molecular mechanisms involved in reducing regenerative capacity with biliary obstruction are suppressed expression of MYC (Tracy et al, 1991), C/EBP, and cyclin E (Nakano et al, 2001). Production of HGF (Hu et al 2003), EGF (Bissig et al, 2000), and IL-6 (Fujiwara et al, 2001) is also altered. Finally, biliary obstruction also impairs enterohepatic circulation and thus negatively affects the regenerative capacity. It is clinically relevant that external biliary drainage for obstructive jaundice markedly suppresses liver regeneration after hepatectomy (Suzuki et al, 1994), whereas internal biliary drainage preserves this capacity (Saiki et al, 1999).
Diabetes Mellitus
Insulin is one of the most important hepatotrophic factors in portal venous blood (Starzl et al, 1975). The binding protein of insulin-like growth factor (IGF), a molecule similar to insulin, also rises substantially after hepatectomy (Demori et al, 2000; Ghahary et al, 1992). Thus, impaired secretion of insulin and IGF in diabetic patients prevents liver regeneration following hepatectomy, as reflected by decreased synthesis of RNA, DNA, and protein on the first postoperative day (Yamada et al, 1977). Enhancement of mitochondrial phosphorylative activity in the remnant liver following hepatectomy is inhibited in proportion to the severity of impaired insulin secretion. Insulin gene transfer via the spleen enhances liver regeneration without causing liver damage and improves nutritional status after hepatectomy in diabetic rats (Matsumoto et al, 2003). Multiple regression analysis in clinical portal vein embolization (PVE) has shown that diabetes mellitus is a risk factor for reduced hypertrophy in the nonembolized lobe (Imamura et al, 1999). These results demonstrate the importance of insulin in hepatic regeneration, and strict glucose control should be aimed for in liver surgery and PVE (Yokoyama et al, 2007a).
Nutritional Status
Hepatic regeneration is metabolically intensive and requires a great deal of energy. Liver regeneration after hepatectomy is associated with a derangement in energy metabolism, measured by a decrease in the ratio of ATP to its hydrolysis product, inorganic phosphate. This depleted energy status is mirrored in biochemical indices of liver function, and restitution parallels the course of restoration of hepatic cell mass (Mann et al, 2002). Nutritional support is undoubtedly the most physiologically sound means to enhance liver regeneration, but there is still little if any clear information regarding the effects of specific nutrients on liver regeneration in humans (see Chapter 24) (Holecek, 1999). From animal models we know that malnutrition is associated with higher postoperative mortality and reduced regeneration following PHx (Skullman et al, 1990
Related posts:
 Preoperative and postoperative nutrition in hepatobiliary surgery
Preoperative and postoperative nutrition in hepatobiliary surgery
 Cytokines in liver, biliary, and pancreatic disease
Cytokines in liver, biliary, and pancreatic disease
 Nonhepatic surgery in the cirrhotic patient
Nonhepatic surgery in the cirrhotic patient
 Liver transplantation for cholangiocarcinoma and other neoplastic diseases
Liver transplantation for cholangiocarcinoma and other neoplastic diseases
 Distal splenorenal shunt
Distal splenorenal shunt
 Interventional endoscopy: Technical aspects
Interventional endoscopy: Technical aspects
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



{kind=link}