Chapter 6 Liver fibrogenesis
Mechanisms and clinical relevance
Liver Fibrogenesis
Liver fibrosis represents a scarring response to either acute or chronic cellular injury. Following acute liver injury, parenchymal cells regenerate to successfully preserve hepatocellular mass and function. This acute process is associated with an inflammatory and fibrogenic response but limited deposition of extracellular matrix (ECM). In contrast, prolonged liver injury leads to sustained production of growth factors, proteolytic enzymes, angiogenic factors, and fibrogenic cytokines. These events culminate in the accumulation of ECM, forming septa that coalesce into broad bands of scar tissue that encircle nodules of hepatocytes and lead to altered microvascular structure (Friedman, 2004). This late stage of fibrosis, termed cirrhosis, ultimately impairs liver function and leads to portal hypertension and its complications (see Chapter 70A).
Progression of fibrosis to cirrhosis typically evolves over decades before clinical events ensue, but disease may progress more rapidly following repeated episodes of severe acute alcoholic hepatitis; subfulminant hepatitis, especially as a result of drug toxicity; and fibrosing cholestasis in patients with hepatitis C virus (HCV) reinfection after liver transplantation (Berenguer et al, 2003). In addition, rapidly progressive acute HCV with fibrosis in men coinfected with human immunodeficiency virus (HIV) has been reported recently (Fierer et al, 2008).
Genetic and environmental factors also influence the course of the disease. For example, polymorphisms in a number of candidate genes involving the inflammatory (TLR4; Guo et al, 2009) or immune response (specific human leukocyte antigen-II [HLA-II] alleles; Powell et al, 2000) may influence the progression of liver fibrosis in humans. Validated groups of single nucleotide polymorphisms (SNPs) can be used to calculate a disease-specific risk assessment for fibrosis progression (HCV: DDXminor allele, DDX-5POLG2 haplotype [Huang et al, 2006]; alcohol-related or chronic cholestatic disorders: TNF-α, interleukin [IL]-1B [Jarvelainen et al, 2001; Donaldson et al, 2001]). These findings have led to the development of a seven-gene signature with good prognostic value in assessing the risk for cirrhosis development in HCV patients (Huang et al, 2007).
The main etiologies of liver fibrosis in Western countries are chronic HCV and HBV infection, alcohol abuse, and nonalcoholic steatohepatitis (NASH; see Chapters 64 and 65). As a generalized tissue response to chronic injury, fibrosis also occurs in many other organs—including the heart, lung, and kidneys—and typically represents the result of ongoing inflammation. Remarkably, up to 45% of all deaths are related to some kind of fibrosis, which emphasizes the importance of this response and explains the growing interest in this field of research (Fig. 6.1).
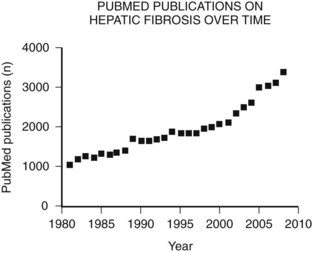
For decades fibrosis was seen as an irreversible disease that progresses to cirrhosis, with a greater risk for developing hepatocellular carcinoma (see Chapters 8C and 80) and liver failure. This meant the only potential treatment for liver fibrosis once it had progressed to cirrhosis was transplantation. Research over the past 30 years has yielded increasing insight into the cellular and molecular mechanisms of this disease, uncovering an orchestrated pathophysiology and identifying the hepatic stellate cell (HSC) as the central cell type in fibrogenesis (Friedman, 1985). Most importantly, this revealed the potential reversibility of this disease and the discovery of potential therapeutic targets.
Molecular and Cellular Mechanisms of Fibrosis
The anatomical arrangement of parenchymal and nonparenchymal cells of the liver contributes to its unique role as an immunity-modulating organ (see Chapter 9) and helps explain how the liver responds to an insult. The liver is composed primarily of epithelial cells, hepatocytes and cholangiocytes, and resident nonparenchymal cells that include hepatic macrophages (Kupffer cells), sinusoidal endothelium, and HSCs. In addition to Kupffer cells, a growing list of specialized immune cells have been characterized, including dendritic cells (DCs), natural killer (NK) cells, and natural killer T (NKT) cells, which indicate that the liver represents a key organ in the regulation of innate immunity.
Common Triggers of Hepatic Fibrogenesis
Ongoing insult to the liver will lead to an increased inflammatory state with activation of hepatic stellate cells, which ultimately tilts the profibrotic–antifibrotic balance toward fibrosis. Ethanol, viral infection, reactive oxygen species (ROS), and bile acids are among the most common stress signals for the liver (Fig. 6.2). An in vitro study further suggests that free fatty acids promote fibrogenesis by indirect activation of HSCs (Wobser, 2009). Less common causes of hepatocellular injury are autoimmune hepatitis, Wilson disease, or hemochromatosis.
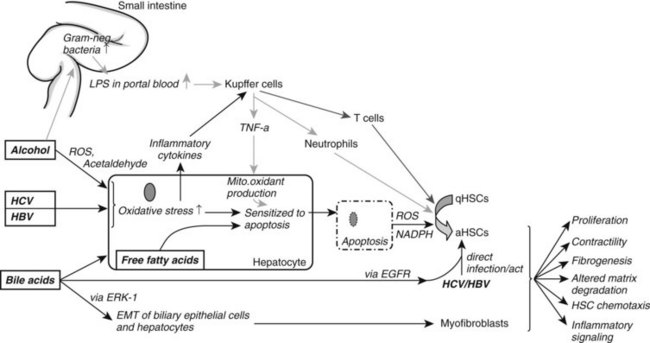
Alcohol decreases gut motility, increases epithelial permeability, and promotes overgrowth of gram-negative bacteria. Consequently, lipopolysaccharide concentration is elevated in portal blood, which activates Kupffer cells through the Toll-like receptor (TLR) 4 complex to generate ROS via reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Wheeler, 2001). Oxidants then upregulate NF-κB in Kupffer cells, which leads to increased tumor necrosis factor (TNF)-α production. TNF-α in turn induces neutrophil infiltration and stimulates mitochondrial oxidant production in hepatocytes, which are then sensitized to undergo apoptosis. Furthermore, acetaldehyde, the main degradation product of alcohol, and ROS both activate HSCs and stimulate inflammatory signals (Maher et al, 1994).
In HCV infection, the virus escapes the HLA-II immune response and infects hepatocytes. This causes oxidative stress, again leading to recruitment of inflammatory cells and HSC activation. HSCs can also be directly activated by either HCV, through membrane receptors (Mazzocca et al, 2005, Schulze-Krebs et al, 2005), or by HBV (Martin-Vilchez et al, 2008).
Bile acids are hepatotoxic agents that typically target hepatocytes, but they may also injure biliary epithelium (Higuchi & Gores, 2003). Damaged hepatocytes turn apoptotic or necrotic, thereby releasing ROS (Nieto et al, 2002) and NADPH oxidase, which both activate HSCs (Canbay et al, 2004a). Injured hepatocytes also release inflammatory cytokines and soluble factors that activate Kupffer cells and stimulate the recruitment of activated T cells. This inflammatory milieu further stimulates the activation of resident HSCs. Bile acids also directly stimulate proliferation of HSCs by activating the epidermal growth-factor receptor (Svegliati-Baroni et al, 2005). In contrast to hepatocytes, HSCs are protected from bile acid–induced apoptosis by excluding bile acids (Svegliati-Baroni et al, 2005).
Nonalcoholic fatty liver disease (NAFLD) is increasingly prevalent as a result of increased obesity in the United States and Western Europe. NAFLD can progress to NASH with consequent fibrosis and cirrhosis (Carter-Kent et al, 2009). The pathogenesis is not fully understood; however, a two-hit model has been proposed with hyperglycemia and insulin resistance representing the first hit, leading to elevated serum levels of free fatty acids. Histologically this correlates to hepatic steatosis. Additional oxidative stress or proinflammatory cytokines are thought to promote hepatocyte apoptosis and recruitment of inflammatory cells, thereby enhancing fibrogenesis.
Hepatic Stellate Cell Activation and Hepatic Myofibroblasts (MFBs)
The HSC has emerged as a central regulator of the liver’s fibrotic and repair responses (Friedman, 2000). In normal liver the HSC is a quiescent cell type that contains cytoplasmic retinoid droplets, representing the major storage site for vitamin A in the body, and expresses the markers desmin and glial fibrillary acidic protein (GFAP; Friedman, 2008).
During liver injury, HSCs undergo activation in response to a range of inflammatory and injury signals (Casini, 1997) produced by damaged hepatocytes and biliary cells; changes in the composition of the ECM; proangiogenic growth factors, like VEGF and angiopoietin; and fibrogenic cytokines that include TGF-β1, angiotensin II, and leptin.
Activation of HSCs is accompanied by loss of retinoid droplets and accumulation of α-smooth muscle actin, a myogenic filament that confers increased cellular contractility. Activated HSCs are characteristically desmin- and α-SMA–positive cells. Highly activated subsets of HSCs are known as hepatic myofibroblasts (MFBs; Friedman, 2008), a cell type that is also characteristic of wound healing in a range of tissues: skin, kidney, lung, bone marrow, and pancreas, among others.
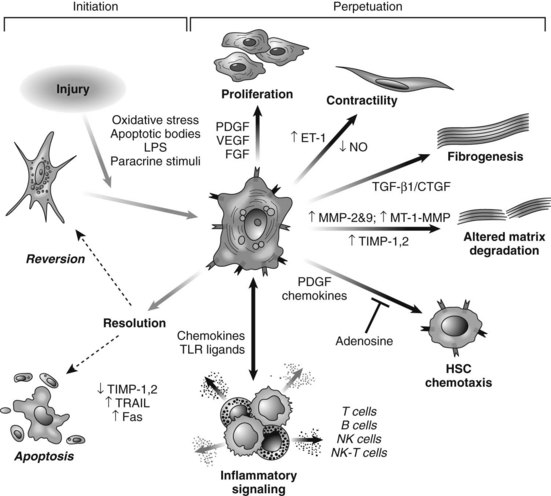
HSC activation can be divided conceptually into two phases: initiation begins with early changes in gene expression and phenotype that result from paracrine stimulation, primarily due to changes in surrounding ECM and exposure to lipid peroxides and products of damaged hepatocytes (Fig. 6.3); perpetuation results from the effects of these stimuli on maintaining the activated phenotype and generating fibrosis. Within the nucleus, a growing number of transcription factors regulate HSC behavior, including peroxisomal proliferator–activated receptors (PPARs), retinoid receptors, NF-κB, JUND, Kruppel-like factor 6 (KLF6), and FOXF1 (Mann & Mann, 2009). A range of general and cell type–specific membrane receptors and signaling pathways also control HSC biology, including receptor tyrosine kinases, chemokine receptors, and integrins (Dodig et al, 2007).
Portal fibroblasts (Beaussier et al, 2007; Ramadori & Saile, 2004) and bone marrow–derived MFBs (Russo et al, 2006) have also been identified as collagen-producing cells in the injured liver. Another emerging source of fibrogenic cells is epithelial-mesenchymal transition (EMT), in which adult hepatocytes of biliary epithelium transdifferentiate into fibrogenic cells (Rygiel et al, 2008). EMT has been extensively characterized in kidney and lung fibrosis, in subsequent animal models and human samples of liver fibrosis, and in the context of hepatocarcinogenesis. Interestingly, key signals regulating EMT also drive activation of HSCs, including TGF-β, RAS, extracellular signal-regulated kinase 1 (ERK1; Zhong et al, 2009), SMAD7, and SHH (Dooley et al, 2008; Lahsnig et al, 2009; Syn et al, 2009).
The relative importance of each cell type in liver fibrogenesis may depend on the origin of the liver injury. In cholestatic liver diseases and ischemia, portal fibroblasts may be especially important (Clouzeau-Girard et al, 2006), whereas hepatic MFBs may be of predominant importance in alcoholic liver disease. In chronic liver injury, progressive recruitment of bone marrow–derived cells may occur over time. However, bone marrow–derived cells may have both fibrogenic and antifibrotic effects (Karlmark et al, 2009). To date, the quantitative contribution to fibrogenesis of nonstellate cell–derived fibroblasts remains unclear.
Functions of Hepatic MFBs
Hepatic MFBs have functions that are distinct from their quiescent cells of origin. They are profibrogenic and promitotic, play a chemotactic and vasoregulatory role, and control the degradation of the ECM (see Fig. 6.3). They also have important immune and phagocytic functions (Friedman, 2008). The regulation of ECM accumulation and degradation by HSCs is reviewed in the next section.
Fibrogenesis
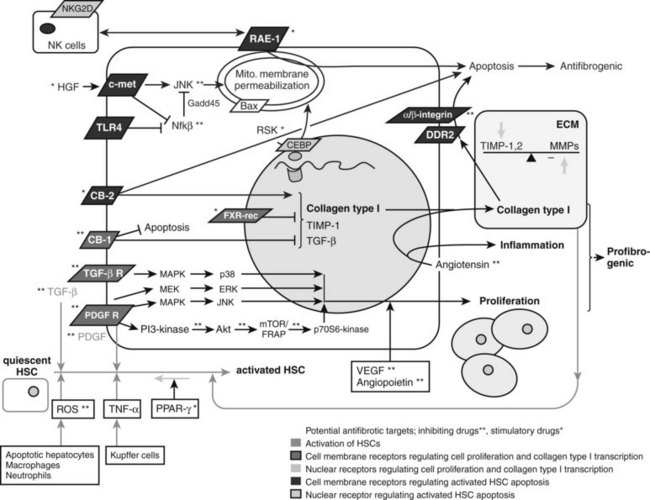
The major profibrogenic signal in the liver is the cytokine TGF-β1 (Bachem et al, 1989a). TGF-β1 is secreted mainly by MFBs (Bissell et al, 1995) but also by platelets (Bachem et al, 1989b) and Kupffer cells (Bilzer et al, 2006). It acts by activating the TGF-βII receptor, which recruits the TGF-βI receptor. SMAD2 and SMAD3 then associate with the TGF-βI receptor (Dooley et al, 2001), are phosphorylated, and recruit SMAD4. This triheteromeric complex then translocates to the nucleus, where it activates profibrogenic transcription factors. TGF-β also activates the MAPK p38 pathway (Cao et al, 2002), which leads to additional, SMAD-independent collagen type 1 synthesis and, in contrast to the SMAD-dependent collagen type 1 synthesis, also leads to a posttranscriptionally regulated stabilization of the collagen type 1 mRNA (Tsukada et al, 2005; Fig. 6.4).
Proliferation
The predominant stimulus to MFB proliferation is the mitogen platelet-derived growth factor (PDGF; Borkham-Kamphorst et al, 2004), in addition to other mitogens that include EGF, VEGF, and FGF. All pathways downstream of PDGF receptor activation promote proliferation. First, JNK is stimulated through MAPK (Schnabl et al, 2001); second, PDGF receptor stimulates the Ras/Raf complex, followed by MEK and ERK engagement (Schnabl et al, 2001); and third, activation of the PI3K pathway leads to Akt activation and phosphorylation of the 70s6 kinase (see Fig. 6.4; Reif, 2003).
Immunoregulation
The liver is a microenvironment of diminished immunogenicity, which is necessary to cope with the high exposure of antigens from the portal vein (Crispe, 2003). This feature also accounts for the tolerance of liver transplantation across ABO barriers and may contribute to the chronic nature of hepatitis B and C, where the virus persists despite the development of an immune response. Upon entry of antigen into the sinusoid, classical antigen-presenting cells (Kupffer cells, dendritic cells) are first encountered. Subsequently, HSCs in the space of Disse may contact antigens. Indeed, HSCs display a wide range of immunoregulatory functions and are an essential part of the local immunogenicity (Gao et al, 2008; Mehal, 2007; see Chapter 9).
Hepatic MFBs produce a range of proinflammatory (e.g., TNF-α; Tiggelman et al, 1995) and antiinflammatory IL-10 cytokines (Tiegs & Lohse, 2009; Safadi et al, 2004) and they recruit lymphocytes through secretion of chemokines (MCP-1, IL-8, CCL21, RANTES; Marra et al, 1998, 1993; Maher et al, 1998), thus amplifying the inflammatory response. However, upon activation, they exert a profound immunosuppressive activity by inducing T-cell apoptosis (Yu et al, 2004). In the setting of liver transplantation, MFB-induced T-cell apoptosis via programmed death ligand-1 (Yu et al, 2004) may enable local immunotolerance of the liver. In liver fibrosis, MFBs may further regulate the contribution of lymphocytes to the course of hepatic fibrosis by ingesting disease-associated lymphocytes (Muhanna et al, 2008).
The interaction between HSCs and immune cells is bidirectional. T cells activate HSCs by IFN-γ, which upregulates both stimulatory (CD80, CD86, CD54) and inhibitory (B7-H1) surface molecules and enhances both inflammatory and suppressive cytokines. However, the inhibitory molecules are thought to override the stimulatory counterparts, resulting in immunosuppression. Lymphocytes can also mediate hepatic fibrosis by activating HSCs. CD8-positive T lymphocytes are more fibrogenic toward stellate cells than CD4-positive T lymphocytes (Safadi et al, 2004). This may explain in part why patients coinfected with HIV and HCV have accelerated fibrosis, as their CD4–CD8 cell ratios are reduced. Of the CD4-positive T lymphocytes, previously called T helper cells, the humoral-mediated immunity by T helper-2 (Th2) cells is profibrogenic in liver injury, whereas the cell-mediated immunity by T helper-1 (Th1) cells via IFN-γ, TNF-α, and IL-2 is antifibrogenic (Shi, 1997).
HSCs can also function as antigen-presenting cells (Winau et al, 2007). They can interact with bacterial lipopolysaccharides directly via TLR4, which amplifies their activation. TLR4 signaling leads to downregulation of a TGF-β pseudoreceptor, BAMBI, which thereby amplifies fibrogenic activity of MFBs (Seki & Brenner, 2008). This might contribute to an accelerated progression of liver cirrhosis, as microbial translocation from the gut to the liver is increased, which is the case in alcoholic liver disease.
Vasoregulation
MFBs play an important role in the regulation of sinusoidal blood flow and may contribute to portal hypertension that is characteristic of advanced liver disease. The release of endothelin-1 (ET-1) can stimulate their contraction through the ETA receptor (Shi-Wen et al, 2004), thereby promoting tissue contraction, increasing portal resistance, and generating portal hypertension. On the other hand, MFBs and endothelial cells also secrete nitric oxide (NO), which is the physiologic antagonist of ET-1.
Structural Features of Hepatic Fibrogenesis
In hepatic fibrosis the total amount of collagen is increased up to sixfold, whereas the parenchymal mass (e.g., hepatocytes) is progressively diminished. The composition of the ECM changes with progression of disease. Collagen type IV in the space of Disse is replaced by interstitial collagen types I and III. Additionally, the discontinuous basal membrane beneath the sinusoidal endothelial cells is replaced by a continuous basement membrane, and sinusoidal fenestrations are reduced. This decreased porosity, also known as capillarization, combined with perisinusoidal fibrosis, scar contraction, and formation of intrahepatic shunts contributes to increased hepatic venous pressure and portal hypertension. Fibrillar collagens produced by MFBs interact with MFBs via discoidin domain receptors and integrins (Olaso et al, 2001), thereby inhibiting apoptosis and increasing MFB proliferation.
With the maturation of the fibrotic scar, not only is the amount of collagen increased, but the scar also becomes increasingly insoluble through chemical cross-linking by transglutaminase, and assembly of collagen fibrils through increased generation of collagen monomers by metalloproteinases from the “ADAM metallopeptidase with thrombospondin type 1 motif”; in addition, metalloproteinase with thrombospondin-type repeats metalloproteinase with thrombospondin type I motif (ADAMTS2; Kesteloot et al, 2007) family. This makes the fibrous septa progressively resistant to proteolysis by matrix metalloproteinases (MMPs). The long-standing clinical dogma—the slower the pace of injury, the less reversible the scar—is supported by animal studies in which even advanced fibrosis of short duration is reversible. Thus, the reversibility of a scar may be limited primarily by the extent of collagen cross linking. Clinically, increased septal thickness and smaller nodule size, both of which reflect more advanced stages of fibrosis, are significant predictors of worse clinical outcomes (Nagula et al, 2006).
Regulation of Collagen Deposition and Degradation
The deposition and degradation of collagen is tightly regulated. MMPs are the key enzymes that degrade fibrillar collagens (types I and III) and noncollagenous ECM substrates. The tissue inhibitor metalloproteinases (TIMPs) are their major antagonists by inactivating proteases and by inhibiting MFB apoptosis (Murphy et al, 2002).
Both decreased levels of interstitial collagenases and increased levels of MMP inhibitors in liver injury create an imbalance that favors reduced degradation of fibrillar collagens in hepatic fibrosis. The interstitial collagenases MMP-1, MMP-8, and MMP-13 in humans and MMP-13 in rodents (Emonard & Grimaud, 1990) unwind the triple-helical collagen type I, which is the principal collagen in the fibrotic liver (Williams & Olsen, 2009), so that each α-chain is presented to the active site of the enzyme (Chung et al, 2004), which cleaves the collagen. Other MMPs (e.g., MMP-2) cannot unwind the triple-helical collagen and thus cannot degrade intact collagen type I alone. In early liver injury, MMP-2 degrades the low-density basement membrane present in the subendothelial space (Zhou et al, 2004a). Its replacement with fibril-forming matrix impairs hepatocyte differentiation and function. During progressive fibrosis, expression of MMP-1 (humans) or MMP-13 (rodents) is decreased, and MMP-2 expression increases (Preaux et al, 1999; Milani et al, 1994). In parallel, the expression of TIMP-1 and TIMP-2, which inhibit the collagen-degrading matrix metalloproteinases, are increased (Kossakowska et al, 1998; Murawaki et al, 1993).
Hepatic macrophages also regulate matrix remodeling (Mitchell et al, 2009), and they are an important source of proteases, including MMP-13 (Hironaka et al, 2000; Fallowfield et al, 2007) in rodents. Macrophages might even be more critical to ECM degradation than HSCs, whose degradation potential is impaired during activation by elevation of the inhibitor TIMP-1. Kupffer cells can further stimulate TIMP-1 expression by hepatic MFBs (Wang et al, 2009), leading to inhibition of MMP activity and also protection of MFBs from apoptosis. In mouse models, macrophages augment fibrogenesis during progression of liver fibrosis, whereas during resolution they hasten matrix degradation through increased production of MMP-13 (Fallowfield et al, 2007).
Although both macrophages and MFBs secrete MMP-1 in humans and MMP-13 in rodents, it is not clear which is the major interstitial collagenase in fibrosis regression, because MMP-1 is only expressed at low levels in liver. Moreover, the cellular origin of those MMPs deemed essential to fibrosis regression remains controversial. Dendritic cells and T lymphocytes are also potential sources, as both can secrete MMP-9 (Chabot et al, 2006; Owen et al, 2003).
Diagnosis and Clinical Monitoring of Hepatic Fibrosis
Many patients with chronic liver disease may initially present with late-stage fibrosis, as earlier stages are often asymptomatic. Thus clinicians must have a high index of suspicion for occult fibrosis, especially in patients with unexplained elevations of liver enzymes, splenic enlargement, stigmata of liver disease, or laboratory or imaging findings suggestive of portal hypertension. When chronic liver disease is suspected, liver biopsy remains the gold standard for diagnosing and staging liver fibrosis, but it is an invasive procedure with risk of adverse events (Bravo et al, 2001; Poynard et al, 2000) and, equally important, a high likelihood of sampling variability (Bedossa et al, 2003) and interpathologist and intrapathologist variability (Bedossa & Poynard, 1996). At least one third of biopsies may deviate by one fibrosis stage between the right and left hepatic lobes in HCV (Regev et al, 2002). Shorter biopsies are associated with an increase in reported diagnoses of mild and moderate fibrosis at the cost of more severe fibrosis, representing an understaging of fibrosis (Colloredo et al, 2003); the smaller the tissue sample, the milder the apparent disease.
Several commonly used histologic staging systems for fibrosis exist. The Histology Activity Index (HAI) score reported by Knodell includes three stages (Knodell et al, 1981), and the Ishak score differentiates six stages, including two stages of cirrhosis, incomplete and complete (Ishak et al, 1995). The METAVIR score, developed by the French METAVIR Cooperative Study Group, is a simple, widely applied five-stage scoring system (Bedossa & Poynard, 1996) that is most commonly used worldwide. It incorporates the fibrosis scores F0 through F4 (F0, no fibrosis; F1, fibrosis without septa; F2, few septa; F3, numerous septa without cirrhosis; F4, cirrhosis) and activity scores A0 through A3 that assess the amount of necroinflammation (A0, no necroinflammatory activity; A1, mild; A2, moderate; A3, severe). All these systems were developed primarily for scoring fibrosis associated with viral hepatitis, with the main emphasis on the degree of necroinflammation. The fewer fibrosis stages within a scoring system, the higher the reproducibility among observers.
Scoring systems have also been specifically developed for different etiologies of liver fibrosis: Ludwig and colleagues proposed a four-stage system to describe fibrosis stages for both primary biliary cirrhosis (Ludwig et al, 1978) and sclerosing cholangitis (Ludwig et al, 1981), and Kleiner and colleagues (Kleiner, 2005) quantified fibrosis in NAFLD with a seven-stage system. The Kleiner stages are stage 0, no fibrosis; stage 1, perisinusoidal or periportal fibrosis (1a: mild, zone 3; 1b: moderate, zone 3; 1c: portal/periportal); stage 2, periportal and perisinusoidal fibrosis; stage 3, bridging fibrosis; and stage 4, cirrhosis.
There is a great need for reliable, quantitative, noninvasive diagnostics of fibrosis, and recent studies indicate steady progress. Cross-sectional imaging studies, such as those provided by computed tomographic (CT) and magnetic resonance imaging (MRI) scans, can reliably demonstrate features of advanced liver disease, including nodularity and signs of portal hypertension (splenomegaly, enlarged caudate lobe, esophageal varices). Diffusion-weighted MRI measures the apparent diffusion coefficient of water, a parameter that depends upon tissue structure, and it has compared favorably to other noninvasive measures for determining advanced fibrosis (Lewin et al, 2007).
Biochemical Tests
The most studied combination serum tests are the AST platelet ratio index (APRI; Wai et al, 2003), the FIB-4 index (Sterling et al, 2006), the Forns test (Forns et al, 2002), and the proprietary FibroTest (Imbert-Bismut et al, 2001). Newer, proprietary biomarker scores include the HepaScore (Adams et al, 2005) and the FibroMeter (Cales et al, 2005). The factors included in the various tests were chosen upon multivariate analysis and are included in Table 6.1
Related posts:
 Preoperative and postoperative nutrition in hepatobiliary surgery
Preoperative and postoperative nutrition in hepatobiliary surgery
 Cytokines in liver, biliary, and pancreatic disease
Cytokines in liver, biliary, and pancreatic disease
 Nonhepatic surgery in the cirrhotic patient
Nonhepatic surgery in the cirrhotic patient
 Liver transplantation for cholangiocarcinoma and other neoplastic diseases
Liver transplantation for cholangiocarcinoma and other neoplastic diseases
 Distal splenorenal shunt
Distal splenorenal shunt
 Interventional endoscopy: Technical aspects
Interventional endoscopy: Technical aspects
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


