Epidemiologic investigations, clinical observations, and family and twin studies in humans, as well as gallstone prevalence investigations in inbred mouse models, support the concept that cholesterol cholelithiasis could result from a complex interaction of environmental factors and the effects of multiple undetermined genes. Quantitative trait locus (QTL) analysis is a powerful genetic method for identifying primary rate-limiting genetic defects and discriminating them from secondary downstream lithogenic effects caused by mutations of the primary genes, and the subsequent positional cloning of such genes responsible for QTLs, followed by the use of manufactured mouse strains with “knockout” or “knockin” of the genes, could lead to the discovery of lithogenic actions of gallstone ( LITH ) genes. The combined use of genomic strategies and phenotypic studies in inbred strains of mice has successfully resulted in the identification of many candidate LITH genes. Because there is exceptionally close homology between mouse and human genomes, the orthologous human LITH genes can be identified from the mouse study. The discovery of LITH genes and more fundamental knowledge concerning the genetic determinants and molecular mechanisms underlying the formation of cholesterol gallstones in humans will pave the way for critical diagnostic and prelithogenic preventive measures for this exceptionally prevalent digestive disease.
Cholesterol cholelithiasis is one of the most prevalent digestive diseases, resulting in a considerable financial and social burden worldwide. In the United States, at least 20 million Americans (12% of adults) are affected. Each year, approximately 1 million new cases are discovered, and more than 700,000 cholecystectomies are performed, making this one of the most common elective abdominal operations. Although many gallstones are silent, approximately one-third eventually cause symptoms and complications. In addition, the unavoidable complications result in 3000 deaths (0.12% of all deaths) annually. In the year 2000, more than 750,000 outpatient clinic visits and more than 250,000 hospitalizations were the result of gallstone-induced gastrointestinal symptoms. As a result, there was a median inpatient charge of $11,584, and medical expenses for the treatment of gallstones exceeded $6 billion. Because the prevalence of gallstones is increasing because of the worldwide obesity epidemic with associated insulin resistance, a key feature of the metabolic syndrome, it is imperative to find potential ways to prevent the formation of gallstones.
During the past 50 years, it has been established that the primary pathophysiologic defect in most cholelithogenic humans is hepatic hypersecretion of biliary cholesterol, which may be accompanied by normal, high, or (less commonly) low secretion rates of biliary bile salts or phospholipids, inducing unphysiological cholesterol supersaturation of gallbladder bile. Recent studies on humans and mouse models have shown that interactions of 5 defects ( Fig. 1 ) result in nucleation and crystallization of cholesterol monohydrate crystals in bile and eventually gallstone formation. These are (1) genetic factors and Lith genes; (2) unphysiological supersaturation with cholesterol due to hepatic hypersecretion of biliary lipids; (3) accelerated phase transitions of cholesterol; (4) dysfunctional gallbladder motility accompanied by immune-mediated gallbladder inflammation, as well as hypersecretion of mucins and accumulation of mucin gel in the gallbladder lumen; and (5) increased amounts of cholesterol from the intestinal source due to high efficiency of cholesterol absorption or slow intestinal motility which aids hydrophobe absorption and augments second bile salt synthesis by the anaerobic microflora. Growth of solid platelike cholesterol monohydrate crystals to form gallstones is a consequence of persistent hepatic hypersecretion of biliary cholesterol together with gallbladder mucin hypersecretion and gel formation and with incomplete evacuation by the gallbladder as a result of its impaired motility function.
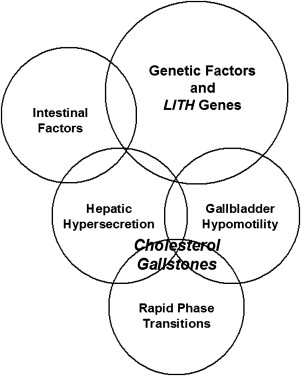
Epidemiologic investigations, clinical observations, and family and twin studies in humans, as well as gallstone prevalence investigations in inbred mouse models, have clearly shown that a complex genetic basis could play a key role in determining individual predisposition to develop cholesterol gallstones in response to environmental factors. A powerful genetic technique, quantitative trait locus (QTL) analysis has been successfully used to identify, localize, and analyze the lithogenic effects of pathophysiologically relevant gallstone ( Lith ) genes in inbred strains of mice. These genetic analyses of cholesterol gallstones in mice have provided many novel insights into the complex pathophysiological mechanisms that are involved in the formation of cholesterol gallstones in humans. On the basis of these studies, a new view has been proposed that hepatic hypersecretion of biliary cholesterol could be induced by multiple Lith genes in mice and humans, as well as insulin resistance as part of the metabolic syndrome, all interacting with cholelithogenic environmental factors to cause the phenotype. This review summarizes recent progress in searching for Lith genes by a strategy of the combined genomic and phenotypic studies in inbred strains of mice, which has been proven to be a powerful approach to understand the complex pathophysiology of this common disease, as well as in elucidating the genetic mechanisms of cholesterol gallstone disease in humans.
QTL study and LITH genes in mice
In 1964, Tepperman and colleagues first established a mouse model of cholesterol gallstones by feeding a special lithogenic diet containing 1% cholesterol and 0.5% cholic acid. Later, Fujihira and colleagues found that at 8 weeks on the lithogenic diet, gallstone prevalence rates varied from 0% to 100% in 7 strains of mice. Alexander and Portman further observed that feeding the lithogenic diet for a longer time, from 8 weeks to 12 weeks, strain differences in gallstone formation still existed in mice. Khanuja and colleagues also found a striking strain difference in gallstone prevalence rates in 9 strains of inbred mice. By using a mouse backcross strategy and QTL analysis, they observed that differences in gallstone susceptibility between gallstone-susceptible C57L/J and resistant AKR/J strains were determined by at least 2 Lith genes, with Lith1 (for lithogenic gene 1) and Lith2 mapping on mouse chromosomes 2 and 19, respectively. These studies show that the mouse is an excellent model for gallstone studies and also permits the investigation of genetics of cholesterol cholelithiasis.
On the basis of these mouse studies, it has been proposed that cholesterol gallstone formation is most likely to result from complex interactions of environmental factors and the effects of multiple, but as yet unknown, genes. Thus, methods for studying genetics of gallstones, a complex genetic trait, could differ from those that are used to discover genes for simple Mendelian defects. To study quantitative polygenic traits, conventional genetic mapping methods are inadequate because they are designed for single-gene traits. As a result, genetic analysis of affected sibling pairs or families is hampered by multiple modes of inheritance of the trait, genetic heterogeneity, and large variations in environmental conditions. It has been proposed that the strategy for identifying the genetic defects underlying the metabolic abnormalities in cholesterol gallstones in inbred strains of mice includes the following steps: (1) QTL analysis in inbred mice with different gallstone prevalence is performed to identify the chromosomal regions that contain Lith genes; (2) all candidate genes in regions that have putative lithogenic effects, and that are colocated with the chromosomal regions of the QTLs, are determined; and (3) the candidate genes are individually evaluated by studying their lithogenic actions in knockout, transgenic, or congenic mice, and, accordingly, their cholelithogenic mechanisms are identified at a molecular level.
The QTL technique provides a useful and powerful method that can be used to identify, locate, and analyze the lithogenic effects of candidate genes in several chromosomal regions. An additional advantage of the QTL technique is that it can be used to discover new genes, whereas knockout and transgenic mice usually provide information only on known genes. The QTL method is used to investigate linkages of the candidate genes to polymorphic genetic markers on the mouse genome. The polymorphic genetic markers are short, variable DNA sequences (ie, microsatellite markers) or even single nucleotides that differ among inbred strains of mice. During the past decades, a systematic mapping of murine genes relevant to the phenotypes of common diseases became possible with the construction of a dense genetic map that consists of thousands of microsatellite markers with known chromosomal localizations. The goal of QTL analysis is to identify the genes underlying these polygenic traits and to gain a better understanding of their lithogenic actions on gallstone formation.
The first QTL analysis of cholesterol gallstones was successfully performed between inbred strains of C57L/J and AKR/J mice. The results from this study showed that (1) gallstone susceptibility in the C57L/J strain is determined by at least 2 unlinked genes; (2) 2 major genes, designated Lith1 and Lith2 , are mapped on mouse chromosomes 2 and 19, respectively; and (3) gallstone formation is determined by a dominant trait. Additional QTLs in other strains of inbred mice with different susceptibility to gallstones have been investigated, and so far, 25 Lith genes have been identified, which are distributed from chromosomes 1 to X, designated Lith1 to Lith25 .
Compared with AKR/J mice, C57L/J mice of both genders displayed biliary cholesterol hypersecretion, early cholesterol supersaturation, accumulation of mucin gel, large gallbladder size, rapid cholesterol crystallization, and early onset and high gallstone prevalence rate. On the lithogenic diet, C57L/J mice failed to downregulate activities of hepatic 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase controlling cholesterol biosynthesis, and cholesterol 7α-hydroxylase and sterol 27-hydroxylase, regulatory enzymes in 2 bile salt synthesis pathways, became profoundly suppressed. The biliary secretion study found that there is absolute and relative biliary cholesterol hypersecretion leading to increased cholesterol saturation index in the face of upregulated bile salt and phospholipid secretion rates in C57L/J mice. C57L/J mice displayed significantly greater absorption of cholesterol from the small intestine, more rapid plasma clearance of chylomicrons and chylomicron remnants, higher activities of lipoprotein lipase and hepatic lipase, greater hepatic uptake of chylomicron remnants, and faster secretion of chylomicron remnant cholesterol from plasma into bile. This finding shows that cholesterol absorbed from the intestine provides an important source for biliary hypersecretion. These data provide important clues for further exploring the roles of Lith genes in pathogenesis of cholesterol cholelithiasis, and for investigating the phenotypes of individual Lith genes.
To confirm whether Lith1 and Lith2 could induce gallstones, congenic strains that carry the C57L/J alleles only in the Lith1 or the Lith2 regions on an AKR/J genetic background were created. Lith1 and Lith2 congenic strains showed gallstone formation comparable with the C57L/J parent, confirming that the Lith1 and the Lith2 QTL regions can cause gallstones. It was found that (1) Lith1 plays a predominant role in determining biliary cholesterol hypersecretion and cholesterol gallstone formation because the gallstone and biliary phenotypes in the Lith1 congenic strain are similar to those in the C57L/J parent; and (ii) Lith2 , but not Lith1 , influences bile salt–independent bile flow.
Candidate genes for cholesterol gallstones in mice
On the basis of the pathophysiology of cholesterol gallstones, Table 1 summarizes major classes of candidate genes for Lith genes, which could have an important effect on gallstone formation in mice. Lithogenic actions of some candidate genes have not yet been identified, and their roles in cholelithogenesis need to be investigated further. Genetic factors contributing to cholesterol gallstones include candidate genes that encode (1) hepatic and intestinal lipid transporters on the apical and basolateral membranes; (2) hepatic and intestinal lipid regulatory enzymes; (3) hepatic and intestinal intracellular lipid transporters; (4) hepatic and intestinal lipid regulatory transcription factors; (5) hepatic lipoprotein receptors and related proteins; (6) nuclear receptors in the liver and small intestine; (7) hormone receptors in the gallbladder; and (8) biliary mucins. Fig. 2 illustrates the locations of Lith genes and the candidate genes on mouse chromosomes.
| Gene Symbol a | Gene Name | Mouse Chr b | cM | Human Ortholog |
|---|---|---|---|---|
| A. Liver | ||||
| (i) Lipid membrane transporters | ||||
| ABCG5 and ABCG8 | ATP-binding cassette sterol transporters G5 and G8 | 17 | 54.5 | 2p21 |
| ABCB4 (MDR2) | Phosphatidylcholine transporter (multiple drug resistance 2) | 5 | 1.0 | 7q21.1 |
| ABCB11 (BSEP, SPGP) | Bile salt export pump (Sister of P-glycoprotein) | 2 | 38.4 | 2q24 |
| ABCC2 (CMOAT, MRP2) | Canalicular multispecific organic anion transporter (multidrug resistance-related protein 2) | 19 | 43.0 | 10q24 |
| SLC10A1 (NTCP) | Sodium/taurocholate cotransporting polypeptide | 12 | 37.0 | 14q24.1 |
| SLC21A1 (OATP1) | Organic anion transporting polypeptide 1 | 6 | A3-A5 | ND |
| SLC22A1 (OCT1, ORCT) | Organic cation transporter 1 | 17 | 7.34 | 6q26 |
| ATP8B1 (FIC1) | Familial intrahepatic cholestasis type 1 | 18 | ND | 18q21-q22 |
| NPC1L1 | Niemann-Pick C1 like 1 protein | 11 | ND | 7p13 |
| (ii) Lipid regulatory enzymes | ||||
| CYP7A1 | Cholesterol 7α-hydroxylase | 4 | ND | 8q11-q12 |
| CYP7B1 | Oxysterol 7α-hydroxylase | 3 | 1.0 | 8q21.3 |
| CYP27A1 | Sterol 27-hydroxylase | 1 | 43.1 | 2q33 |
| HMGCR | 3-Hydroxy-3-methyglutaryl-coenzyme A reductase | 13 | 49.0 | 5q13.3-q14 |
| SOAT2 (ACAT2) | Sterol O -acyltransferase 2 (acyl-coenzyme A:cholesterol acyltransferase 2) | 15 | 61.7 | 12q13.13 |
| (iii) Intracellular lipid transporters | ||||
| CAV1 | Caveolin 1 | 6 | A2 c | 7q31.1 |
| CAV2 | Caveolin 2 | 6 | A2 | 7q31.1 |
| FABP1 | Fatty acid-binding protein 1, liver | 6 | 30.0 | 2p11 |
| NPC1 | Niemann-Pick type C1 | 18 | 4.0 | 18q11-q12 |
| OSBP | Oxysterol-binding protein | 19 | 7.0 | 11q12-q13 |
| PCTP | Phosphatidylcholine transfer protein | 11 | 52.0 | 17q21-q24 |
| SCP2 | Sterol carrier protein 2 | 4 | 52.0 | 1p32 |
| (iv) Lipid regulatory transcription factors | ||||
| NR2B1 (RXRα) | Retinoid X receptor α | 2 | 17.0 | 9q34.3 |
| NR1H3 (LXRα) | Liver X receptor α | 2 | 40.4 | 11p11.2 |
| NR1H2 (LXRβ) | Liver X receptor β | 7 | ND | 19q13.3 |
| NR1H4 (FXR) | Farnesoid X receptor | 10 | 50.0 | 12q23.1 |
| NR1C1 (PPARα) | Peroxisomal proliferator activated receptor α | 15 | 48.8 | 22q13.31 |
| NR1C2 (PPARδ) | Peroxisomal proliferator activated receptor δ | 17 | 13.5 | 6p21.2-p21.1 |
| NR1C3 (PPARγ) | Peroxisomal proliferator activated receptor γ | 6 | 52.7 | 3p25 |
| NR0B2 (SHP) | Small heterodimer partner | 4 | 60.0 | 1p36.1 |
| NR1I3 (CAR) | Constitutive androstane receptor | 1 | 92.6 | 1q23.3 |
| SREBF1 | Sterol regulatory element binding transcription factor 1 | 11 | ND | 17p11.2 |
| SREBF2 | Sterol regulatory element binding transcription factor 2 | 15 | ND | 22q13 |
| SCAP | SREBF cleavage activating protein | 9 | 58.0 | 3p21.3 |
| ESR1 (ERα) | Estrogen receptor α | 10 | 12.0 | 6q25.1 |
| ESR2 (ERβ) | Estrogen receptor β | 12 | 33.0 | 14q23.2 |
| (v) Lipoprotein receptors and related genes | ||||
| APOB100 | Apolipoprotein B100 | 12 | 2.0 | 2p24-p23 |
| APOE | Apolipoprotein E | 7 | 4.0 | 19q13.2 |
| LRP1 | LDLR-related protein 1 | 10 | B2-D1 | 12q13-q14 |
| LRP2 (GP330) | LDLR-related protein 2 (Megalin, Glycoprotein 330) | 2 | 40.0 | 2q24-q31 |
| LDLR | Low density lipoprotein receptor | 9 | 5.0 | 19p13.3 |
| SRB1 | Scavenger receptor class B type I | 5 | 68.0 | 12q24.31 |
| LPL | Lipoprotein lipase | 8 | 33.0 | 8p22 |
| LIPC (HPL) | Hepatic lipase | 9 | 39.0 | 15q21-q23 |
| LCAT | Lecithin cholesterol acyltransferase | 8 | 53.0 | 16q22.1 |
| LRPAP1 | LRP-associated protein 1 | 5 | 20.0 | 4p16.3 |
| PLTP | Phospholipid transfer protein | 2 | 93.0 | 20q12-q13.1 |
| B. Gallbladder | ||||
| (i) Hormone receptors | ||||
| CCK | Cholecystokinin | 9 | 71.0 | 3p22-p21.3 |
| CCK1R (CCKAR) | Cholecystokinin-1 receptor (CCK-A receptor) | 5 | 34.0 | 4p15.1-p15.2 |
| ESR1 (ERα) | Estrogen receptor α | 10 | 12.0 | 6q25.1 |
| ESR2 (ERβ) | Estrogen receptor β | 12 | 33.0 | 14q23.2 |
| PGR | Progesterone receptor | 9 | ND | 11q22-q23 |
| (ii) Mucin | ||||
| MUC1 | Mucin 1 | 3 | 44.8 | 1q21 |
| MUC3 | Mucin 3 | 5 | 75 | 7q22 |
| MUC4 | Mucin 4 | 16 | ND | 3q29 |
| MUC5AC | Mucin 5ac | 7 | 69.0 | 11p15.5 |
| MUC5B | Mucin 5b | 7 | 69.0 | 11p15.5 |
| MUC6 | Mucin 6 | 7 | 69.0 | 11p15.5 |
| (iii) Lipid membrane transporters | ||||
| ABCG5 and ABCG8 | ATP-binding cassette sterol transporters G5 and G8 | 17 | 54.5 | 2p21 |
| SRB1 | Scavenger receptor class B type I | 5 | 68.0 | 12q24.31 |
| (iv) Lipid regulatory enzymes | ||||
| HMGCR | 3-Hydroxy-3-methyglutaryl-coenzyme A reductase | 13 | 49.0 | 5q13.3-q14 |
| SOAT2 (ACAT2) | Sterol O -acyltransferase 2 (acyl-coenzyme A:cholesterol acyltransferase 2) | 15 | 61.7 | 12q13.13 |
| C. Small intestine | ||||
| (i) Lipid membrane transporters | ||||
| ABCG5 and ABCG8 | ATP-binding cassette sterol transporters G5 and G8 | 17 | 54.5 | 2p21 |
| SRB1 | Scavenger receptor class B type I | 5 | 68.0 | 12q24.31 |
| SLC10A2 (IBAT) | Ileal (apical) sodium/bile salt transporter | 8 | 20 | 13q33 |
| (ii) Lipid regulatory enzymes | ||||
| HMGCR | 3-Hydroxy-3-methyglutaryl-coenzyme A reductase | 13 | 49.0 | 5q13.3-q14 |
| SOAT2 (ACAT2) | Sterol O -acyltransferase 2 (acyl-coenzyme A:cholesterol acyltransferase 2) | 15 | 61.7 | 12q13.13 |
| (iii) Intracellular lipid transporters | ||||
| CAV1 | Caveolin 1 | 6 | A2 | 7q31.1 |
| CAV2 | Caveolin 2 | 6 | A2 | 7q31.1 |
| FABP6 (ILLBP) | Fatty acid-binding protein 6, ileal | 11 | 24.0 | 5q33.3-q34 |
| OSBP | Oxysterol-binding protein | 19 | 7.0 | 11q12-q13 |
| SCP2 | Sterol carrier protein 2 | 4 | 52.0 | 1p32 |
| NPC1L1 | Niemann-Pick C1 like 1 protein | 11 | ND | 7p13 |
| APOB48 | Apolipoprotein B48 | 12 | 2.0 | 2p24-p23 |
| APOAI | Apolipoprotein AI | 9 | 27.0 | 11q23-q24 |
| APOAIV | Apolipoprotein AIV | 9 | 27.0 | 11q23 |
| APOCIII | Apolipoprotein CIII | 9 | 27.0 | 11q23.1-q23.2 |
| (iv) Lipid regulatory transcription factors | ||||
| NR2B1 (RXRα) | Retinoid X receptor α | 2 | 17.0 | 9q34.3 |
| NR1H3 (LXRα) | Liver X receptor α | 2 | 40.4 | 11p11.2 |
| NR1H2 (LXRβ) | Liver X receptor β | 7 | ND | 19q13.3 |
| NR1H4 (FXR) | Farnesoid X receptor | 10 | 50.0 | 12q23.1 |
| NR1C1 (PPARα) | Peroxisomal proliferator activated receptor α | 15 | 48.8 | 22q13.31 |
| NR1C2 (PPARδ) | Peroxisomal proliferator activated receptor δ | 17 | 13.5 | 6p21.2-p21.1 |
| NR1C3 (PPARγ) | Peroxisomal proliferator activated receptor γ | 6 | 52.7 | 3p25 |
| ESR1 (ERα) | Estrogen receptor α | 10 | 12.0 | 6q25.1 |
| ESR2 (ERβ) | Estrogen receptor β | 12 | 33.0 | 14q23.2 |
Related posts:
 Targets for Current Pharmacologic Therapy in Cholesterol Gallstone Disease
Targets for Current Pharmacologic Therapy in Cholesterol Gallstone Disease
 Diagnosis and Management of Gallbladder Polyps
Diagnosis and Management of Gallbladder Polyps
 Surgical Treatment of Gallstones
Surgical Treatment of Gallstones
 LithGenes and Genetic Analysis of Cholesterol Gallstone Formation
Management of Gallbladder Cancer
Diagnosis and Management of Gallbladder Polyps
LithGenes and Genetic Analysis of Cholesterol Gallstone Formation
Management of Gallbladder Cancer
Diagnosis and Management of Gallbladder Polyps
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


