Ischemic Enteritis
When the blood supply to a tissue is interrupted, a sequence of chemical events leads to cellular dysfunction, edema, and ultimately cell death. Tissue anoxia results in anaerobic metabolism and lactic acidosis. Fewer high-energy bonds are created, and the cell is deprived of the
energy needed to maintain homeostasis (182) (Fig. 6.92). Ischemic injury forms a continuum of changes that range from increased mucosal permeability to frank necrosis. The clinical severity of ischemic enteritis varies widely from massive and sometimes fatal hemorrhagic infarctions to silent transient minimal ischemic episodes. Vascular occlusion and periods of hypotension or vasoconstriction account for most cases of intestinal ischemia. The ischemia may therefore be caused by diseases intrinsic or extrinsic to the bowel. Small intestinal ischemic necrosis predominantly affects elderly individuals with underlying cardiovascular disease. However, intestinal ischemia may affect individuals of any age, including infants. Intestinal ischemia complicates peripheral vascular disease, various vasculopathies, some infections, intussusception and torsion, and certain drug therapies.
energy needed to maintain homeostasis (182) (Fig. 6.92). Ischemic injury forms a continuum of changes that range from increased mucosal permeability to frank necrosis. The clinical severity of ischemic enteritis varies widely from massive and sometimes fatal hemorrhagic infarctions to silent transient minimal ischemic episodes. Vascular occlusion and periods of hypotension or vasoconstriction account for most cases of intestinal ischemia. The ischemia may therefore be caused by diseases intrinsic or extrinsic to the bowel. Small intestinal ischemic necrosis predominantly affects elderly individuals with underlying cardiovascular disease. However, intestinal ischemia may affect individuals of any age, including infants. Intestinal ischemia complicates peripheral vascular disease, various vasculopathies, some infections, intussusception and torsion, and certain drug therapies.
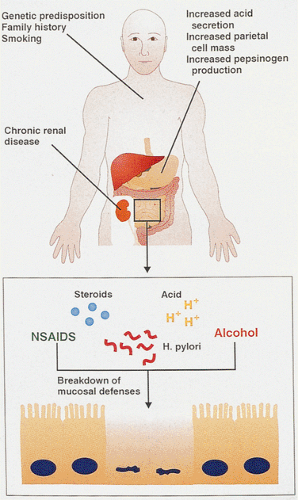 FIG. 6.88. The pathogenesis of duodenal ulcer disease is multifactorial and involves host factors as well as environmental factors. Most patients exhibit increased acid secretion, increased parietal cell mass, and increased pepsinogen production. Environmental agents such as steroids, nonsteroidal anti-inflammatory drugs (NSAIDs), alcohol, or Helicobacter pylori further contribute to the injury by breaking down mucosal defenses. |
All forms of ischemic damage (see Table 6.5) share the underlying feature that the blood supply fails to meet the local tissue demands required to fulfill normal functions and/or maintain normal structure. Prolonged cessation of blood flow inevitably results in cell death because of the diminished delivery of oxygen and metabolic substrates and the accumulation of potentially cytotoxic end products of anaerobic metabolism. Small intestinal blood supply has to be reduced by >50% to induce detectable tissue injury (183). The extent and duration of the ischemia depends on several factors, including the nature of the intestinal vasculature, luminal bacterial virulence, and the duration of the ischemic episode. The first detectable sign of ischemic injury is increased capillary permeability. With continuing ischemia, detectable epithelial cell injury occurs. Mucosal cells are shed at an increased rate, and damage to the plasma membrane of unshed cells results in leakage of cytoplasmic digestive enzymes and tryptic digestion of the mucosa. Arteriolar spasm and decreased perfusion pressure accentuate the extent of the ischemic damage. Necrosis and subsequent bacterial invasion develop when the mucosal barrier becomes defective. The basic pathologic response to ischemia is mucosal coagulative necrosis. If reflow is re-established, acute inflammation develops.
No single process represents the critical event in ischemic injury. Depletion of cellular energy stores and accumulation of toxic metabolites both contribute to cell death. Re-establishing the blood flow (reperfusion) is required to reverse the injury because it allows cellular regeneration and washout of toxic metabolites. However, reperfusion of ischemic tissues also paradoxically injures the tissues (Fig. 6.93) (184). In fact, most of the injury that occurs in intestinal ischemia occurs during the reperfusion period due to the production of reactive oxygen metabolites by activated neutrophils and other inflammatory cells. The severity of reperfusion injury depends on the duration of the preceding hypoxia, with it being more severe following partial rather than total intestinal ischemia (185). The sudden reintroduction of oxygen into the anoxic tissues unleashes oxygen-free radical cascades that overwhelm endogenous defenses. Many derive from the hypoxanthine-xanthine oxidase system (186). Superoxide and hydrogen peroxide increase mucosal and vascular permeability, recruit and activate neutrophils, and act as the precursors of more damaging hydroxyl radicals via the Fenton and myeloperoxidase reactions (Fig. 6.93) (186). Aggressive luminal factors (such as pancreatic proteases, especially trypsin) contribute to the mucosal damage and potentiate bacterial translocation and sepsis.
Neutrophil–endothelial cell interactions are a prerequisite for ischemic microvascular injury (187). The hypoxia induces endothelial cells to produce various adhesion molecules, including integrins, members of the immunoglobulin superfamily, and selectins. These powerful chemoattractants
and chemoactivators attract leukocytes and platelets to reperfused sites and promote their adherence, transendothelial migration, and activation. As a result, one sees a massive mucosal influx of neutrophils. The infiltrating neutrophils are also a major source of reactive oxygen metabolites (ROMs), including O2–, H2O2, OH·, HOCl, and certain n-chloramines.
and chemoactivators attract leukocytes and platelets to reperfused sites and promote their adherence, transendothelial migration, and activation. As a result, one sees a massive mucosal influx of neutrophils. The infiltrating neutrophils are also a major source of reactive oxygen metabolites (ROMs), including O2–, H2O2, OH·, HOCl, and certain n-chloramines.
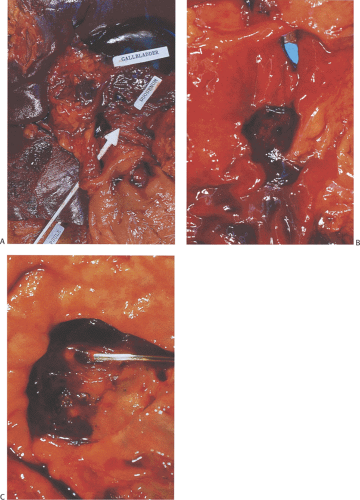 FIG. 6.89. Gross appearance of peptic duodenal ulcers. A and B derive from the same specimen dissected at the time of autopsy. The patient had numerous ulcerations involving the stomach, esophagus, and duodenum. The plastic arrow is inserted through a perforating duodenal ulcer. B: Higher magnification of a different ulcer showing the presence of a granular base. The specimen illustrated in A and B is from a patient with Zollinger-Ellison syndrome. C: Image from a different patient showing the presence of a large visible vessel containing a probe. |
Acute mesenteric infarctions result from mesenteric arterial occlusion (Fig. 6.94), nonocclusive low-flow states, mesenteric vein thrombosis (Fig. 6.95), or vasculitis.
Mesenteric artery thrombosis and embolization (Figs. 6.96 and 6.97) occur with about equal frequency. Thrombi almost invariably overlie atheromatous plaques occupying the proximal few centimeters of the vessel. In contrast, emboli lodge at bifurcation points or in a distal branch. Ischemia also complicates numerous other conditions that ultimately obliterate the intestinal vascular supply (Table 6.5).
Mesenteric artery thrombosis and embolization (Figs. 6.96 and 6.97) occur with about equal frequency. Thrombi almost invariably overlie atheromatous plaques occupying the proximal few centimeters of the vessel. In contrast, emboli lodge at bifurcation points or in a distal branch. Ischemia also complicates numerous other conditions that ultimately obliterate the intestinal vascular supply (Table 6.5).
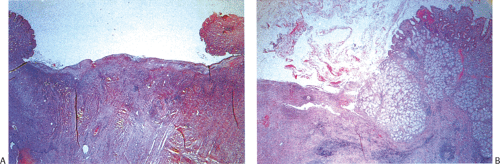 FIG. 6.90. Duodenal ulcer. A: An ulcer with typical overhanging margins is seen. It has the same zones as gastric peptic ulcers (see Chapter 4). B: Ulceration extends deep into the duodenal wall. Brunner gland hyperplasia is seen. |
TABLE 6.5 Causes of Intestinal Ischemia | |
|---|---|
|
Ischemia in Low Flow States (Nonocclusive Ischemia)
Low flow states usually result from decreased cardiac output following primary cardiac disease (infarction or arrhythmia), hypovolemia, shock, vascular shunting, or a combination of low mesenteric flow and mesenteric arterial vasoconstriction. Patients who develop low-flow states are typically elderly and they often have concomitant atherosclerotic vascular disease. Even though the blood supply to the superficial mucosa is fairly well maintained during shock, hypoxic injury still develops within 1 to 2 hours. Several pathogenic mechanisms account for the ischemic necrosis including vasoconstriction with increased resistance to blood flow, redistribution of the blood flow away from the mucosa, increased capillary filtration via relaxation of the precapillary sphincter smooth muscle fibers, and intestinal countercurrent mechanisms in the villi that shunt oxygen away from the villous tips (Fig. 6.98) (188). This explains why the villous tips become anoxic first and explains why early and minimal injury always occurs first at the villous tip (Fig. 6.99).
Arterial Occlusive Disease
Arterial occlusive disease occurs secondary to narrowing, thrombosis, embolism, or hemorrhage under an atheromatous plaque. Occlusions also result from aortic aneurysms, aortic dissections, obstruction by mural thrombi, and tumors that externally compress the vessels. The occlusion may involve one or all of the intestinal arterial trees. Atheromatous occlusion progresses slowly enough for collateral circulation to develop, and as a result patients often have severe disease involving all the mesenteric arteries before they become symptomatic. Intestinal infarction secondary to atheromatous occlusion of a single vessel is rare (189). (About 50% of patients over the age of 50 have atheromatous
narrowing or occlusion of the celiac axis.) The disease is most likely to be most severe in patients with diabetes. The superior mesenteric artery is affected more commonly than the inferior mesenteric artery. The most severe atherosclerotic lesions affect the proximal 2 cm of the superior and inferior mesenteric arteries. Embolic occlusion accounts for one third of cases of mesenteric vascular occlusion (190). Massive, acute, and often fatal embolism usually results from migration of an intracardiac mural thrombus complicating heart disease. Cholesterol emboli migrate from aortic
plaques, especially following catheterization, resulting in localized or widespread intra-abdominal ischemia. Valvular endocarditis may shed small mycotic emboli. Intestinal damage develops within minutes of total circulatory arrest.
narrowing or occlusion of the celiac axis.) The disease is most likely to be most severe in patients with diabetes. The superior mesenteric artery is affected more commonly than the inferior mesenteric artery. The most severe atherosclerotic lesions affect the proximal 2 cm of the superior and inferior mesenteric arteries. Embolic occlusion accounts for one third of cases of mesenteric vascular occlusion (190). Massive, acute, and often fatal embolism usually results from migration of an intracardiac mural thrombus complicating heart disease. Cholesterol emboli migrate from aortic
plaques, especially following catheterization, resulting in localized or widespread intra-abdominal ischemia. Valvular endocarditis may shed small mycotic emboli. Intestinal damage develops within minutes of total circulatory arrest.
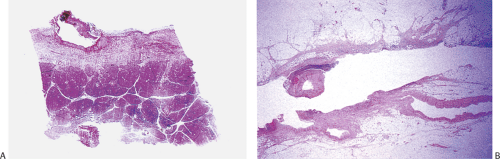 FIG. 6.91. Penetrating duodenal ulcer. A: The bottom of an ulcer bed has eroded a large extraintestinal vessel. The structure at the base is the pancreas, the periphery of which has become fibrotic due to the inflammatory response. B: Erosion into the serosal fat, exposing several branches of a major vessel. |
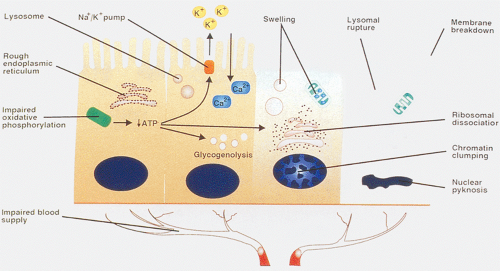 FIG. 6.92. Intestinal ischemia. Intestinal ischemia develops when insufficient arterial blood reaches the intestinal mucosa through the presence of a thrombus, an embolus, an atheromatous plaque, a vascular spasm, low-flow states, or obstruction of venous outflow. As a result, oxidative metabolism becomes impaired, leading to decreased adenosine triphosphate (ATP), increased glycolysis, acidosis, and changes in nuclear chromatin, as well as in other cellular organelles. The decreased ATP also leads to decreased functioning of the sodium-potassium pump with an influx of calcium ions and water and an efflux of potassium. As a result, the cells swell, as do intracellular organelles. As the organelles become damaged, ribosomes decrease and protein synthesis decreases, interfering with reparative processes. |
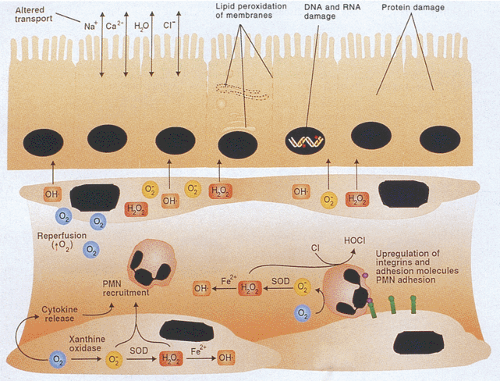 FIG. 6.93. During ischemia, cellular adenosine triphosphate (ATP) is converted to adenosine monophosphate and further catabolized to hypoxanthine, which serves as an oxidizable substrate for xanthine oxidase. The enzyme xanthine oxidase (XO) is the rate-limiting enzyme in nucleic acid degradation. XO generates H2O2 and O2– during the oxidation of hypoxanthine or xanthine. Free radicals are also generated via the Fenton reaction, especially during cell reperfusion. The free radicals recruit polymorphonuclear leukocytes (PMNs) to the reperfused area. The neutrophils adhere to the endothelial cells secondary to increased transcription of integrins and adhesion molecules on both neutrophils and endothelial cells. Free radicals also diffuse into the local tissues, causing abnormalities in the enterocytes with lipid peroxidation of membranes and damage to DNA, RNA, and proteins, and alterations in cellular transport mechanisms. |
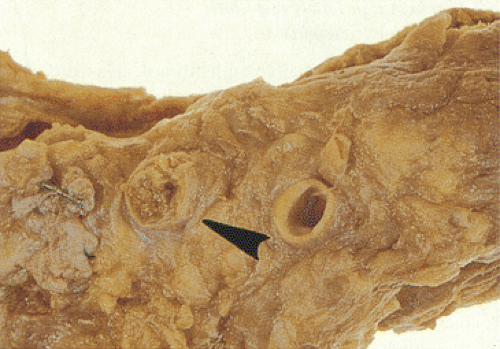 FIG. 6.94. Thrombosed superior mesenteric artery. The arrowhead points to the orifice of the superior mesenteric artery as it exits the aorta. |
Sudden occlusion of the superior mesenteric artery produces hemorrhagic infarction in the area supplied by the occluded vessel (as modified by the presence of a collateral blood supply). The infarcted area may extend from the proximal jejunum to the transverse colon, producing a pattern of ischemic enterocolitis (36). However, usually a small area is involved, and there is a sharp dividing line between the normal and ischemic parts of the bowel (Fig. 6.100).
Generally, one has little difficulty in diagnosing patients with acute, diffuse, transmural ischemic necrosis. These patients abruptly become symptomatic. The most common manifestation is poorly localized colicky abdominal pain that becomes constant and unremitting as the disease progresses. The pain results from spasm of the muscularis
propria. Diarrhea develops and stools become overtly bloody. As the ischemic muscle loses its contractile functions, much of the spasm ceases. The patient then experiences abdominal tenderness, positive rebound signs, and evidence of peripheral circulatory collapse. At this point, the bowel is usually beyond recovery and requires surgical resection. With further disease progression, the abdomen distends and bowel signs disappear. As the ischemia persists and infarction develops, the patients often develop an elevated white blood count, fever, and signs of peritonitis.
propria. Diarrhea develops and stools become overtly bloody. As the ischemic muscle loses its contractile functions, much of the spasm ceases. The patient then experiences abdominal tenderness, positive rebound signs, and evidence of peripheral circulatory collapse. At this point, the bowel is usually beyond recovery and requires surgical resection. With further disease progression, the abdomen distends and bowel signs disappear. As the ischemia persists and infarction develops, the patients often develop an elevated white blood count, fever, and signs of peritonitis.
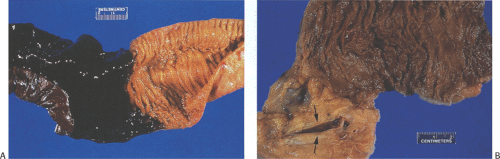 FIG. 6.95. Intestinal infarction secondary to portal vein thrombosis. A: Gross photograph of the junction of the infarcted intestinal segment with normal mucosa. There is a sharp line of demarcation between the infarcted tissue (black tissue on the left) and the viable, noninfarcted tissue on the right. B: Dissection through the peri-intestinal fat showing a branch of the portal venous system, which has been opened and contains a long thrombus (arrows). |
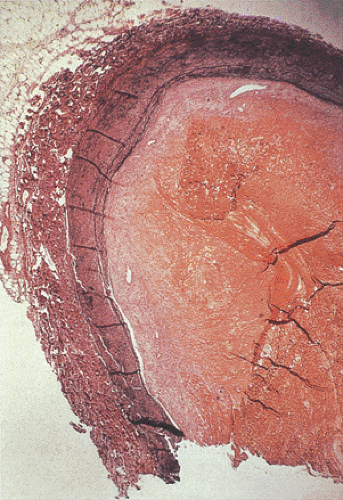 FIG. 6.96. Histologic section through the portal vein showing the presence of an organized thrombus. |
Mesenteric Venous Thrombosis
Mesenteric venous thrombosis is a relatively rare disease primarily affecting the elderly in their 6th and 7th decades of life (191). The prevalence of mesenteric vein thrombosis ranges from 0.003% in the general hospital population to 0.05% of autopsied populations (192). Venous thrombosis accounts for 5% to 15% of all cases of mesenteric ischemia and infarction (Fig. 6.101) (193). Multiple etiologic factors may exist in any one patient. For example, a patient requiring splenectomy may have a pre-existing abnormality involving the coagulation system, experience intraoperative trauma to regional veins, and develop a transient thrombocytosis caused by the splenectomy. Its presence in younger persons suggests that the patient has a hypercoagulable state. Twenty-five to fifty percent of cases have no predisposing cause; these are classified as primary venous thrombosis. Regardless of the cause of the thrombosis, egress of blood from the intestine becomes impaired, causing the mesenteric arterial pressure to rise and arterial blood flow to slow, leading to the development of ischemia. Increased intraluminal pressure further interferes with mucosal viability.
Approximately 95% of all mesenteric thromboses involve the superior mesenteric vein and lead to ischemia or infarction of the small bowel or proximal colon (191). In a small number of cases, the thrombosis develops over an extended period, permitting the development of collateral venous drainage from the involved intestinal segments. Depending on the cause, mesenteric venous thrombosis may begin in the portal vein and extend back into the mesenteric vein and its branches, or it may begin in smaller peripheral mesenteric venous branches and proceed up into the portal vein.
Propagating portal vein thrombosis causes portal hypertension. Thrombi often arise in the distal arcades and propagate proximally.
Propagating portal vein thrombosis causes portal hypertension. Thrombi often arise in the distal arcades and propagate proximally.
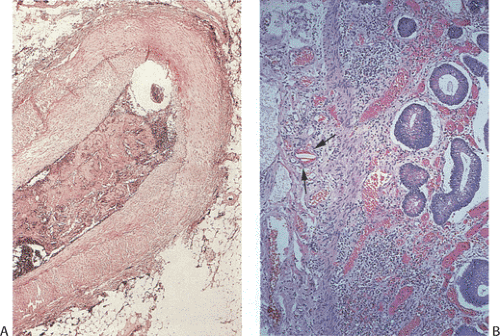 FIG. 6.97. Atheromatous emboli. A: Cross section through the superior mesenteric artery demonstrating the presence of a large atheromatous embolus. B: Emboli extend into the smaller vessels just beneath the muscularis mucosae (arrows). |
Patients present with a constellation of nonspecific findings. The clinical presentation is characterized by gradually increasing colicky abdominal pain. With increased blockage of the collaterals by new thromboses, the patient develops nausea and vomiting, an acute abdomen, and rectal bleeding. At this time, surgical intervention is required. Involvement of the splenic or portal vein with a propagating thrombus may
result in portal hypertension (191). Patients with venous occlusion tend to have a subacute course producing days or weeks of abdominal pain. Bloody ascites is common.
result in portal hypertension (191). Patients with venous occlusion tend to have a subacute course producing days or weeks of abdominal pain. Bloody ascites is common.
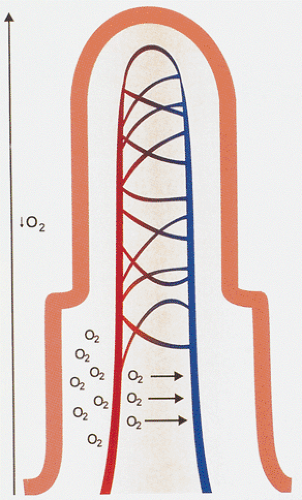 FIG. 6.98. Diagram of countercurrent mechanism demonstrating shunting that occurs at the villous base. It diverts oxygen from the villous tips to the base of the crypts during anoxic periods. |
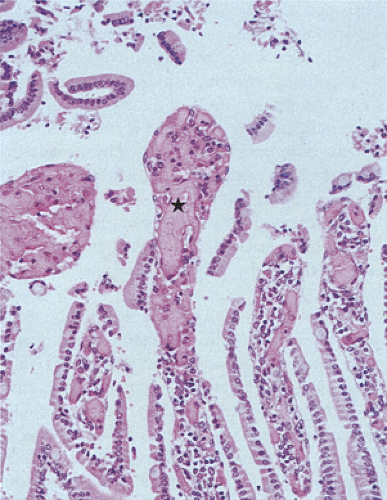 FIG. 6.99. Early ischemia demonstrating marked capillary congestion and loss of epithelium from the villous tips. One of these denuded villus tips is indicated by the star. Taken in isolation, the changes may resemble those seen in autolysis. No reperfusion has occurred and therefore acute inflammatory cells are absent. |
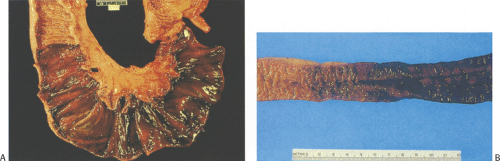 FIG. 6.100. Small bowel ischemia. A: Hyperemic infarcted areas with adjacent friable pink-tan mucosa. B: Area of infarction with gradual hyperemia of adjacent lesser involved mucosa. |
The diagnosis of mesenteric venous thrombosis is usually made during laparotomy for an acute abdomen. At the time of surgery, the serosal and mucosal surfaces appear mottled, hemorrhagic, and discolored with fibrinous exudates deposited on them. The bowel appears thinned in areas of transmural hemorrhagic infarction. Adjacent areas show patchy ischemia. The mesentery usually appears thickened, hemorrhagic, and edematous, and it contains numerous cordlike thrombosed veins. The arteries usually appear normal. Numerous recent, organizing, and partially recanalized thrombi are observable in the mesenteric venous vasculature, and the bowel wall shows intramural hemorrhage and variable degrees of edema and ischemic necrosis with ulceration and acute inflammation. The thrombosis occurs in the absence of phlebitis. The arteries remain uninvolved. Patients may develop Budd-Chiari syndrome.
Mechanical Obstruction of Venous Return
Venous return becomes impeded when the bowel undergoes torsion, volvulus, or intussusception, or when it becomes strangulated and herniates. External compression of the vasculature causes obstruction of the relatively thin-walled and low-pressure venous system before the arterial supply is affected. As a result, the bowel becomes congested with blood, hemorrhagic, and edematous. Ischemic necrosis then develops rapidly. The histologic features resemble those of mesenteric venous thrombosis, except that thrombi are absent.
Gross Features of Ischemic Injury
The pathologic features of intestinal ischemia are similar, no matter what the underlying cause. Ischemia characteristically appears segmental in nature. Early on, the ischemic bowel is edematous and pale with submucosal congestion, hemorrhage, and focal mucosal sloughing (Fig. 6.102). As the disease progresses, the serosa becomes dusky and purple or dark red due to the presence of large amounts of intraluminal blood. The serosa loses its normal glistening appearance and appears dull. The mucosa appears necrotic, nodular, and ulcerated. Extensive submucosal hemorrhage may be present. In early lesions, only the mucosa and sometimes the submucosa are affected; the muscularis propria usually remains normal. As the necrosis progresses, all of the bowel wall layers become damaged and the serosa becomes purplish green. With increasing damage, the intestinal wall thins and becomes friable and membranous exudates form. If the ischemia results from mesenteric venous thrombosis, one sees old and new thrombi extruding from the veins. Ulcers may be present that may be superficial or deep. Perforation may develop. Patients with chronic damage may have mural fibrosis and strictures.
Histologic Features of Ischemic Injury
The histologic features of ischemia depend on whether the changes are acute and minimal or whether they result from transmural infarction. They also depend on whether there is a total vascular occlusion without reperfusion or whether the blood flow is merely reduced below the local needs and reflow has occurred. Therefore, ischemic lesions vary from patchy congestion and ulceration to extensive infarction, gangrene, and perforation.
Most pathologists encounter small intestinal ischemia when they examine large resected segments of necrotic bowel. In this situation, it is easy to diagnose the ischemia. Diagnostic difficulties only occur when one encounters early lesions or when complications develop. Biopsies to establish a diagnosis of ischemia or to rule out other etiologies of enterocolitis are much less common than colonic biopsies for the same purposes. Therefore, the biopsy features of intestinal ischemia are extensively discussed in Chapter 13.
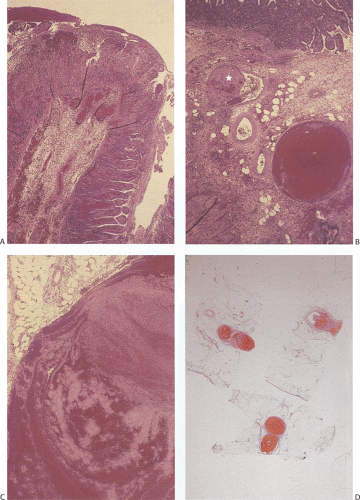 FIG. 6.101. Portal vein thrombosis in a patient with protein S deficiency. The histologic features derive from the specimen illustrated in Figure 6.95. A: The patient developed ischemia with reperfusion injury. There is glandular dropout, loss of villous epithelium, villous congestion, and inflammation. B: Higher magnification of the submucosa showing the presence of a congested and a thrombosed (star) submucosal vein. C: Higher magnification of one of the vessels showing the lines of Zahn in the thrombus. D: The mesenteric fat contains numerous thrombosed branches of the portal vascular system. |
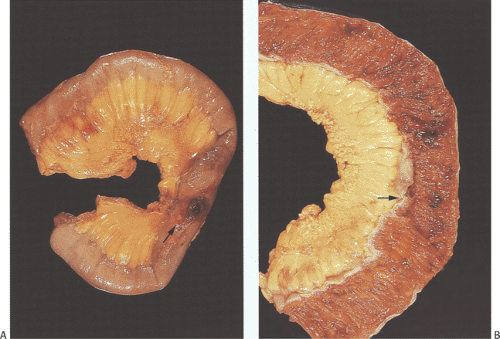 FIG. 6.102. Gross features of intestinal ischemia. A: Unopened specimen with a perforation (arrow). B: Opened specimen showing a localized area of ischemia (arrow). |
Because the mucosa is the most vulnerable part of the intestinal wall, it is damaged first (Fig. 6.103). Early epithelial damage results from loss of energy-dependent processes, causing intercellular edema, and epithelial detachment. Membrane-enclosed cytoplasmic blebs develop on the basal side of the enterocytes where they attach to the basement membrane. This begins the epithelial detachment process. This process starts before the enterocytes display signs of irreversible damage. The process advances from the villous tips to the crypt bases. With more severe or more prolonged ischemia, or both, the epithelium lining the sides of the villi lifts off the basement membrane until the epithelial cells are lost completely (Fig. 6.104). Within 1 hour of total vascular occlusion the upper two thirds of the villi becomes denuded. Then, the villous core disintegrates. The crypt cells often remain intact with little histologic evidence of damage. In
other cases, one sees crypt hypoplasia–villous atrophy interfering with normal cellular turnover. Occasionally, crypt dilation becomes prominent. Later, the epithelial cells become markedly attenuated and the crypts appear compressed and atrophic as the lamina propria swells and hemorrhages. After 5 hours of total acute occlusion, almost the entire intestinal wall appears necrotic.
other cases, one sees crypt hypoplasia–villous atrophy interfering with normal cellular turnover. Occasionally, crypt dilation becomes prominent. Later, the epithelial cells become markedly attenuated and the crypts appear compressed and atrophic as the lamina propria swells and hemorrhages. After 5 hours of total acute occlusion, almost the entire intestinal wall appears necrotic.
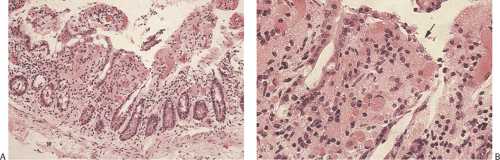 FIG. 6.103. Ischemic enteritis. A: Medium magnification showing loss of superficial epithelial lining from the villi. Large numbers of the crypts are identifiable only by residual cells at the base of the glands. B: Higher magnification of the superficial area showing edema, vascular congestion, and degeneration and sloughing of the epithelium from the tips (arrow). |
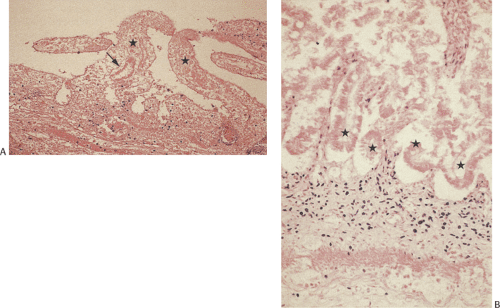 FIG. 6.104. Intestinal infarction. A: The small intestine is completely necrotic with transmural coagulative necrosis. The ghosts of the preexisting villous cores are present (stars). Ghosts of dead enterocytes are indicated by the arrow. There is no inflammation because reperfusion did not occur. B: Coagulative necrosis associated with anoxia. Sections of several crypts are indicated by the stars. The epithelium is sloughing and the nuclei lack their typical basophilia. This photograph shows the increased sensitivity of the epithelium to anoxic effects. The underlying stroma still contains intact nuclei in the stromal cells. |
In cases of total occlusion, the capillaries appear congested with red cell extravasation and coagulation necrosis unaccompanied by acute inflammation. The pathologic findings are definitive but mimic autolysis because of the absence of neutrophilic infiltrates. Longer periods of ischemia eventually destroy the crypt bases and the underlying musculature. Overt perforation occurs when the ischemic process involves the entire thickness of the bowel wall. The presence of fibrin thrombi in mucosal capillaries serves as a useful diagnostic feature of ischemia (Fig. 6.105), but it only becomes evident once epithelial breakdown occurs. In severe injury the crypts drop out completely, and if the damage heals the area becomes fibrotic. The degree of fibrosis that develops depends on the extent of the ischemic damage.
If partial blood flow is maintained, then the effects of the reperfusion are superimposed on the ischemic damage. Early stromal changes consist of edema of the lamina propria, often associated with hemorrhage and emigration of neutrophils through the epithelial surface, especially at the villous tips. There is often prominent telangiectasia. A pseudomembrane composed of necrotic epithelium, fibrin, and inflammatory cells develops (Fig. 6.106). In cases of severe injury, the ischemic process extends into the submucosa and muscularis propria and serosa (Fig. 6.106). If the ischemia only affects a small segment, collateral circulation may allow healing to begin even as the infarction is proceeding at a smaller focus,
resulting in regenerative changes superimposed on degenerative ones.
resulting in regenerative changes superimposed on degenerative ones.
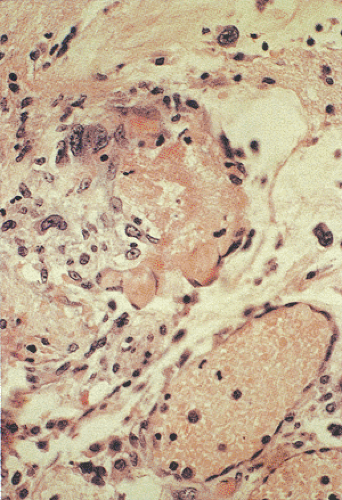 FIG. 6.105. Ischemia with focal fibrin thrombi in the vasculature. |
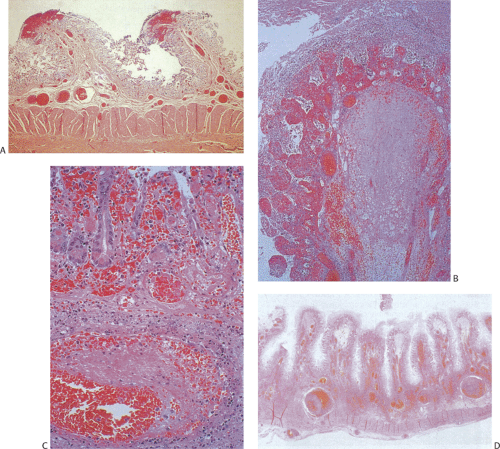 FIG. 6.106. Acute ischemia. A: Early hemorrhagic necrosis of the tips of the folds is seen. The epithelium is extensively denuded. B: Extensive hemorrhagic infarction of the tip of the folds. The entire mucosal structure is completely infarcted. A pseudomembrane covers the surface of the bowel. The submucosal structures are edematous and hemorrhagic. C: Marked necrosis is seen in the small bowel. The mucosa appears hemorrhagic and telangiectatic. An organizing thrombus is seen in the underlying blood vessel. D: Severe extensive transmural infarction of the small bowel.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|





