CHAPTER 107 Infectious Enteritis and Proctocolitis and Bacterial Food Poisoning
In Western countries, diarrhea leads to high morbidity with loss of time from school and work. Overall, physicians in the United States are consulted annually for 8.2 million diarrheal episodes.1,2 A long list of complications, some mild and others life-threatening, can accompany infectious diarrhea. Medical costs and loss of productivity resulting from infectious diarrhea amount to more than $23 billion each year in the United States.
Our knowledge of infectious diarrheal disease has expanded exponentially in the past two decades. References 3 to 6 are excellent discussions of various aspects of enteric infections.3–6
SUSCEPTIBILITY TO INTESTINAL INFECTION
HOST DEFENSE FACTORS
Gastric Acidity
Ingested pathogenic bacteria and other pathogens first must survive passage through the stomach to infect the small or large intestine. In this regard, gastric acidity is the first line of defense.7 Most bacterial pathogens are highly susceptible to low pH, and thus exposure to gastric acid significantly reduces the number of viable bacteria after their ingestion. Gastric juice with a pH of less than 4.0 is rapidly bactericidal, whereas bacteria survive prolonged exposure to gastric juice from patients who are achlorhydric.8 In experimental studies of cholera in healthy adults, clinical infection did not develop when as many as 1010 cholera bacteria were ingested, whereas as few as 104 Vibrio cholerae were able to produce disease when organisms were administered with sodium bicarbonate9; even fewer bacteria were necessary to produce clinical illness when organisms were directly instilled into the duodenum. Naturally occurring cholera also occurs more often in achlorhydric patients.10 The gastric barrier also may be important in preventing other enteric infections such as salmonellosis11 and shigellosis.12
Intestinal Motility
Organisms surviving the milieu of the stomach enter the small intestine where normal propulsive motor activity clears them. Some bacteria, including Clostridium difficile, Clostridium perfringens, and heat-stable enterotoxin-producing Escherichia coli, elaborate toxins that impair intestinal motility.13 In experimental animals it is often necessary to restrict intestinal motility with pharmacologic agents or with ligatures to allow enteropathogens to establish infection.14
Intestinal Microflora
The normal intestinal microflora, primarily in the colon, resist colonization of the intestine by newly introduced bacteria. Products elaborated by the resident microflora, including lactic acid and short-chain fatty acids, are toxic to many bacterial pathogens, and when the intestinal microflora are altered in instances such as administration of an antibiotic, colonization resistance is lessened and the host may be more susceptible to intestinal infection (e.g., C. difficile).15 Alteration of intestinal flora by antibiotics also increases susceptibility to salmonellosis.16,17
Mucus
Mucus, in concert with intestinal motility, provides a physical barrier to bacterial proliferation and mucosal colonization. Gastric mucus can act in conjunction with gastric acidity as the first line of enteric defense. Differences in the carbohydrate composition of intestinal mucus between immature and mature rats suggest that this difference plays a role in the reduced host defense of the immature animal.18
Systemic and Local Immune Mechanisms
The mucosa’s antibacterial immune response is quite complex and important in combating enteric pathogens. Secretory antibody in the intestine appears before serum antibody does in response to intestinal infection with Shigella.19 In cholera, there is a better correlation between the level of coproantibody and immune protection than there is with serum antibody and resistance to enteric infection with this pathogen20; however, both mucosal and systemic immune systems provide important protection against pathogenic bacteria. These immune responses may be directed against multiple targets. For example, the immune response against cholera may be directed against the toxin or the bacterium and originate from either the mucosal immune system (secretory IgA) or from the serum (IgG). Regardless, both serum and secretory antibodies exert their protective effects at the intestinal level, even though the serum components are produced outside the intestine.
Others
Breast-feeding also serves as a defense mechanism against bacterial enteropathogens. Breast-fed infants are less susceptible to bacterial diarrhea than are formula-fed infants.21,22 Multiple factors are responsible for this protection. Breast milk contains secretory IgA antibodies against specific enteropathogens that survive passage through the infant’s gastrointestinal tract.23 Other components such as lactoferrin, lysozyme, and lactoperoxidase also have anti-infective properties, and breast milk glycolipids can interfere with toxin or microbial adherence.24
BACTERIAL FACTORS
Adherence
The ability of bacteria to adhere to host mucosal cells is a critical virulence factor in enterotoxin-producing and invasive bacteria as well as in enteroadherent E. coli (EAEC) and enteropathogenic E. coli (EPEC). Bacterial adherence to host mucosal cells may be the predominant virulence factor, as in the case of EPEC; one of two important factors (adherence plus toxin elaboration), as in the case of enterotoxigenic organisms; or only one of several factors required for expression of full pathogenicity, as seen with invasive organisms. Bacteria that cause disease by adhesion alone do not elaborate any of the traditional enterotoxins, but rather they adhere tightly to the mucosa of both the small and large intestine.25 The classic EPEC and the EAEC26 are typical of this group. Other organisms, including enterotoxigenic E. coli (ETEC) and the invasive Salmonella and Shigella species, also must adhere to the intestinal surface to be fully pathogenic.
Studies on the mechanism by which EPEC cause diarrhea show that they attach to the intestinal mucosa in a characteristic manner, producing ultrastructural changes known as attaching-effacing lesions27 that lead to elongation and destruction of microvilli.25,28 This pattern of bacterial binding to enterocytes also has been referred to as attaching and effacing adherence27 and the particular morphologic alteration as pedestal formation (Fig. 107-1).25
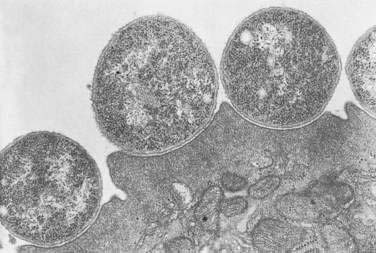
The laboratory counterpart of mucosal colonization is adherence in tissue culture to various cell lines such as Hep-2 and HeLa. A characteristic form of localized adherence is observed only with classic EPEC serotypes. These events occur in the following three phases29:
The presence of a plasmid in EPEC serves to increase intimin production; this process is needed for localized adherence to occur.31 EPEC strains with localized adherence produce acute diarrhea when these strains are administered to normal volunteers.32 The role of the eaeA gene as a virulence factor in human EPEC infection has been confirmed in volunteer challenge studies.33
Enterotoxigenic organisms also require expression of bacterial adherence for proliferation of the organisms and colonization, as well as for full expression of toxicity.34 ETEC adhere to the surface of the small bowel epithelium without penetrating the epithelial layer and do so by mechanisms different from those used by EPEC. The most important mechanism by which enterotoxigenic bacteria adhere to the intestinal mucosa is related to specific protein antigens on the surface of the bacterial cell known as pili or fimbriae, also referred to as adherence antigens or colonization factor antigens.35 These pili bind to specific receptor sites on the surface of the intestinal cell via specific ligand-receptor interactions.
Evidence that these colonization factors (e.g., pili and lectins) are important to the pathogenesis of E. coli diarrheal disease in animals is derived from the observations by Moon36 that loss or gain of fimbriae by genetic manipulation results in the loss or gain of the ability to adhere to and colonize the intestine. Adherence not only permits colonization but also can facilitate the delivery of enterotoxin to the epithelium and might even enhance the ability of the organism to elaborate enterotoxin.34,36
Enterotoxin Production
Enterotoxins are polypeptides, secreted by bacteria, that alter intestinal salt and water transport without affecting mucosal morphology.5,37,38 Many organisms elaborate enterotoxins (e.g., V. cholerae, Shigella, ETEC, and Staphylococcus aureus), and several enterotoxins may be elaborated by a single organism. Although most enterotoxins affect the small intestine, the colon also may be a target organ.
Whether the enterotoxin is ingested preformed or is first expressed within the intestinal lumen, the toxin-enterocyte or toxin-colonocyte interaction begins with the binding of the enterotoxin to a specific mucosal receptor. The toxin-receptor interaction increases the concentration of an intracellular mediator, resulting in alteration of salt and water flux. Thus far, three intracellular mediator systems have been shown to be involved in the pathogenesis of enterotoxigenic diarrhea: adenylate cyclase and cyclic adenosine monophosphate (cAMP), the guanylate cyclase and cyclic guanosine monophosphate (cGMP) systems, and intracellular calcium.39,40 Alterations in these mediator systems have similar effects on transport processes to decrease the coupled influx of sodium and chloride and to stimulate the active secretion of chloride from the cell into the intestinal lumen. Other intracellular mediator systems involved in the pathogenesis of bacterial diarrhea include protein kinase C and arachidonic acid metabolites, among others.
Cholera toxin (molecular weight ≈84,000 kDa), which stimulates adenylate cyclase, is a prototypical enterotoxin (Fig. 107-2 and Chapter 99), the toxin-enterocyte interaction of which is well understood. Cholera toxin is composed of an A subunit surrounded by five B subunits that bind the toxin to a ganglioside (GM1) receptor on the brush border membrane of the villus epithelial cell. The A subunit, which consists of two parts (A1 and A2) linked by a disulfide bond, slowly penetrates the brush border membrane and is cleaved into its two component peptides. Reduction of this bond releases the active A1 peptide that traverses the cell to the basolateral membrane, where it stimulates the ribosylation of Gs, the stimulatory subunit of a heterotrimeric G protein. This action results in the irreversible activation of Gs and an increase in cytosolic cAMP. This cAMP in turn activates cAMP-dependent kinases that inhibit NaCl-coupled transport and stimulate chloride secretion.
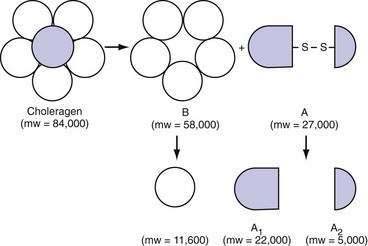
Figure 107-2. Subunit structure of cholera toxin (choleragen) (see text). mw, molecular weight.
(From Fishman PH. Action of cholera toxin: Events on the cell surface. In Field M, Fordtran JS, Schultz SG, editors. Secretory Diarrhea. Bethesda, Md: American Physiological Society; 1980. p 86.)
In addition to cholera toxin, other important enterotoxins are those elaborated by E. coli.41,42 Two classes of E. coli enterotoxins are known: heat-labile (LT) and heat-stable (ST) toxins. LT, which exists in two forms (LT-1 and LT-2),42 is a large molecular-weight protein that causes diarrheal disease similar to, but less severe, than, cholera. The subunit structure and mechanism of action of LT-1 and LT-2 also are similar to those of cholera toxin; although cholera toxin and LT bind to a glycolipid receptor, specifically GM1, an additional glycoprotein receptor might exist for LT.42 Heat-stable enterotoxins (STas) also may be elaborated by E. coli and bind to brush border receptors on enterocytes and colonocytes, a receptor guanylate cyclase, to increase intracellular levels of cGMP. STb is an unrelated enterotoxin elaborated by some E. coli pathogenic for pigs. Other organisms that elaborate highly homologous heat-stable enterotoxins include Yersinia enterocolitica, Citrobacter, and non-O1 vibrios.42
Cytotoxin Production
Cytotoxins are polypeptides that cause cell injury, inflammation, intestinal secretion through inhibition of protein synthesis or via a cascade involving one or more inflammatory mediator substances, and cell death. Examples of organisms that produce cytotoxins include C. difficile (see Chapter 108),15 some EPEC, enterohemorrhagic E. coli (EHEC), and Shigella.43 The mechanisms by which cytotoxins cause cell injury, inflammation, and intestinal secretion are numerous and complex and include inhibition of protein synthesis, disruption of cellular actin and tight junction integrity, mitochondrial damage, and adenosine triphosphate depletion among others.
Mucosal Invasion
The mechanism of mucosal invasion involves invasion of enterocytes or colonocytes by the infecting organisms with subsequent intracellular multiplication, resulting in cell injury and possibly cell death. Shigella species are classic examples of invasive enteropathogens. Salmonella species, Campylobacter jejuni, Y. enterocolitica, and some (enteroinvasive) strains of E. coli invade intestinal cells and pass into the lamina propria, where they elicit an inflammatory response and cause mucosal ulceration.4,6
Unlike enterotoxigenic organisms that favor colonization of the small intestine, invasive organisms primarily, but not exclusively, colonize the colon. In salmonellosis, the ileum is colonized in addition to the colon; in shigellosis, the small intestine is colonized transiently early in the course of the disease when watery diarrhea rather than dysentery is the predominant symptom.44 Subsequently, colonization occurs in the colon and bloody diarrhea ensues. In the cases of Shigella and Salmonella species, the ability to invade the gastrointestinal mucosa is of primary importance in establishing the enteric infection.45,46
Bacterial invasion alone is not sufficient to establish disease; other properties of invading organisms also are required. In the case of Shigella species, the organisms must multiply intracellularly. Thus, strains of Shigella flexneri that can invade but cannot multiply do not cause disease when fed to a susceptible host.45 Intracellular multiplication of Shigella organisms also involves lateral spread to adjacent intestinal cells and their death. In the cases of Salmonella species and Y. enterocolitica, however, the organisms penetrate into the lamina propria and can disseminate to extraintestinal sites. As a consequence of mucosal invasion and of the intramucosal multiplication of the organisms, an acute inflammatory reaction develops and mucosal ulceration can occur. Gross ulceration of the colonic mucosa commonly occurs in shigellosis, which accounts for dysenteric stools, but it is much less common with Salmonella and Yersinia infections. Yersinia infection more commonly manifests with microscopic and minute ulcerations involving both the ileum and colon.
CLASSIFICATION OF BACTERIAL DIARRHEA
DIAGNOSIS OF INFECTIOUS DIARRHEAL DISEASE
EVALUATION OF THE PATIENT
The initial step in the diagnostic evaluation of a patient with acute diarrhea should be a thorough history and physical examination, the goals of which are to identify patients who may be at risk of severe illness or susceptible to complications and those who will benefit from specific therapy. Most patients simply need rehydration therapy. Consideration of the patient’s general health, severity and duration of illness, and the setting in which the illness was acquired should enable the clinician to determine who needs further evaluation (Fig. 107-3).
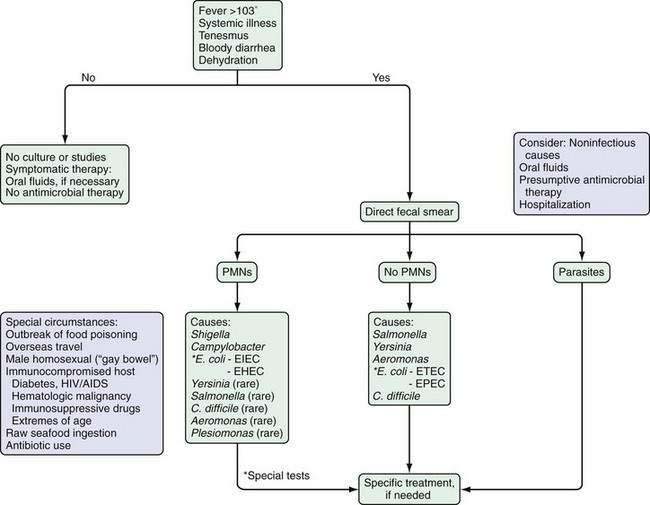
Patients who are debilitated, malnourished, or immunocompromised and those who have severe comorbid illnesses are at increased risk for complications of diarrhea and infection. They can require hospitalization and early diagnostic tests. Other patients who also require a more-aggressive approach include those with systemic signs and evidence of an inflammatory diarrhea, illness lasting more than three to four days, a history or physical examination suggesting a disease process that will benefit from specific therapy, and infection with certain specific organisms (Table 107-1).6
Table 107-1 Pathogens Indicating Need for Antimicrobial Therapy in Patients with Infectious Diarrhea
| Bacterial Infection |
| Protozoal Infection |
| Sexually Transmitted Diseases |
From Park SI, Giannella RA. Approach to the adult patient with acute diarrhea. Gastroenterol Clin North Am 1993;22:483.
Because the number of conditions that cause acute and chronic diarrhea is large, a useful method is to classify the diarrheal illness into one of two clinical syndromes: a watery, noninflammatory diarrheal syndrome and an inflammatory diarrheal syndrome (Table 107-2); a subgroup of the latter is the proctitis diarrheal syndrome. Categorization into one of these syndromes limits the number of potential causes and diagnostic tests that need to be considered. This classification can usually be made on clinical grounds and with simple, inexpensive diagnostic tests.
Table 107-2 Characteristics That Help Distinguish Inflammatory from Noninflammatory Diarrhea
| CHARACTERISTIC | INFLAMMATORY DIARRHEA | NONINFLAMMATORY DIARRHEA |
|---|---|---|
| Clinical presentation | Bloody, small-volume diarrhea; lower quadrant cramps; patients may be febrile and toxic | Large-volume, watery diarrhea; patients may have nausea, vomiting, cramps |
| Site of involvement | Colon | Small intestine |
| Diagnostic evaluation | Indicated | Indicated only if the patient is severely volume depleted or appears toxic |
| Fecal leukocytes | Present | Absent |
| Causes | Shigella spp., Salmonella spp., Entamoeba histolytica, Campylobacter spp., Yersinia spp., invasive Escherichia coli, Clostridium difficile | Viruses, Vibrio spp., Giardia lamblia, enterotoxigenic E. coli, enterotoxin-producing bacteria, food-borne gastroenteritis |
From Park SI, Giannella RA. Approach to the adult patient with acute diarrhea. Gastroenterol Clin North Am 1993; 22:483-97.
Inflammatory Diarrhea
Patients with inflammatory diarrhea usually present with numerous small-volume stools that may be mucoid, grossly bloody, or both. Such patients may appear toxic and usually are febrile. Abdominal cramping may be severe. Because of the small stool volumes, these patients are less likely to be dehydrated than those with noninflammatory diarrhea. Physical findings might point to a specific diagnosis (Table 107-3).
Table 107-3 Clinical Finding(s) That Suggest the Causative Organisms for Some Inflammatory Diarrheas
| FINDING | CAUSATIVE ORGANISMS |
|---|---|
| Hemolytic-uremic syndrome | Shigella spp., enterohemorrhagic E. coli |
| Reactive arthritis | Salmonella spp., Shigella spp., Campylobacter spp., Yersinia spp. |
| Peritoneal signs | C. difficile, enterohemorrhagic E. coli |
| Right lower quadrant tenderness | Yersinia spp. |
| Thyroiditis, pericarditis, glomerulonephritis | Yersinia spp. |
From Park SI, Giannella RA. Approach to the adult patient with acute diarrhea. Gastroenterol Clin North Am 1993; 22:483.
Organisms causing inflammatory diarrheas usually affect the colon and either invade or elaborate cytotoxins, resulting in an acute inflammatory reaction with disruption of the epithelial barrier, mucus, red blood cells, and white blood cells in the stool (Table 107-2). Microbes causing this syndrome include Shigella, Campylobacter, EHEC, C. difficile, Salmonella, Yersinia, and Entamoeba histolytica. Fecal leukocytes (or positive stool lactoferrin test) indicate an acute inflammatory process, and sheets of polymorphonuclear leukocytes (PMNs) usually indicate colitis. The acute inflammatory diarrheal syndrome also can have an noninfectious etiology, for example, ulcerative colitis, Crohn’s disease, radiation or ischemic colitis, and diverticulitis. Table 107-4 lists the organisms that may be associated with the presence of fecal leukocytes.47
Table 107-4 Fecal Leukocytes in Intestinal Infections
| Usually Present |
| Present or Absent |
| Usually Absent |
DAEC, diffusely adhering Escherichia coli; EAEC, enteroaggregative E. coli; EHEC, enterohemorrhagic E. coli; EIEC, enteroinvasive E. coli; EPEC, enteropathogenic E. coli; ETEC, enterotoxigenic E. coli.
LABORATORY DIAGNOSIS
After taking an adequate medical history and performing a physical examination, the clinician should be able to classify the acute diarrheal illness as inflammatory or noninflammatory; to determine its severity and whether complications are likely; and to determine if diagnostic testing is needed and, if so, which tests should be used. In general, most episodes of acute diarrheal illness in the United States are self-limited; diagnostic testing may be kept to a minimum, and treatment is aimed at preventing dehydration. Investigations should be performed only if it is likely the result will influence management and outcome. Diagnostic testing should be reserved for patients with severe illness, including large-volume dehydrating diarrhea, severe abdominal pain, or a prolonged course (i.e., longer than three days); patients with bloody stools, systemic symptoms such as fever higher than 101°F, or prostration; patients with history of recent travel to high-risk areas; and patients at high risk for complications because of extremes of age, diabetes mellitus, and immunocompromise such as acquired immunodeficiency syndrome (AIDS), hematologic malignancy, or use of immunosuppressive medications (see Fig. 107-3).
Fecal Leukocytes
A particularly useful technique to focus the differential diagnosis is microscopic examination of the stool for PMNs (see Table 107-4). Invasive pathogens that primarily affect the colon, such as Shigella and Campylobacter, produce a “sea of polys,” as well as red blood cells. The toxigenic organisms, viruses, and food-poisoning bacteria cause a watery stool that harbors few formed elements. Stool tests for lactoferrin or calprotectin (proteins made by PMNs) in fecal specimens are available and provide a rapid and sensitive alternative to microscopy for identifying PMNs and, by inference, inflammatory diarrhea.48
Stool Cultures
Stool cultures are ordered too frequently. In most microbiology laboratories, routine stool cultures are processed for Shigella, Salmonella, and Campylobacter. Other enteric pathogens such as Yersinia, Vibrio, and E. coli O157:H7 are not sought routinely. Therefore, if clinical suspicion is high, the microbiology department needs to be notified to search for these pathogens. Because of sporadic shedding of pathogens (nontyphoidal Salmonella spp., Salmonella typhi) and because most episodes of acute diarrhea are caused by viruses, undetectable pathogens, or noninfectious causes, stool cultures are not usually positive. At Massachusetts General Hospital, the isolation rate of bacterial pathogens from 2000 fecal cultures in 1980 was 2.4%.49 In patients with severe diarrhea requiring hospitalization, the bacterial isolation rate from feces is somewhat higher, ranging from 27% to 43%,50,51 and up to 58% in a study that used more-advanced techniques.52 Even in patients hospitalized for dysentery, the rate of positivity for microbiologic diagnosis is only 40% to 60%. In community patients with severe acute gastroenteritis (more than four fluid stools per day, lasting at least three days and with at least one associated symptom), the yield of a stool culture and ova and parasite examination increased to 88%.53 In outbreaks of gastroenteritis in the United States, only one half of the cases have a confirmed etiology, of which two thirds are bacterial in origin. These figures suggest that many cases of acute diarrhea are caused by unidentified pathogens.
TOXIGENIC PATHOGENS
VIBRIO CHOLERAE
Microbiology
Toxigenic V. cholerae that agglutinates in O1 antiserum is the main cause of epidemic cholera. There are two major biotypes of V. cholerae O1: classic and El Tor. The latter strain is responsible for the current pandemic that began in 1961. El Tor vibrios are somewhat hardier than others in nature. Clinical disease is similar with both biotypes, although on average, El Tor infections are milder. The major serotypes associated with clinical disease are Inaba and Ogawa; a rare third type is Hikojima. The El Tor Inaba type was responsible for the 1991 outbreak in South America. There also are unique O1 cholera strains (e.g., V. parahaemolyticus and V. vulnificus) that cause endemic disease along the Gulf Coast of the United States.54
A newly described toxigenic non-O1 strain, now designated V. cholerae O139 Bengal, was responsible for an epidemic that started in southern India and Bangladesh in late 1992 and spread rapidly to many countries in Southeast Asia.55,56 This strain was classified as a new serogroup because it did not react with antisera to the previously identified 138 serogroups.56
Cholera Toxin
All wild strains of V. cholerae, including O139, elaborate the same enterotoxin, a protein molecule with a molecular weight of 84,000 kDa (see Fig. 107-2).57 The structural genes for the cholera toxin are encoded by a filamentous bacteriophage.58 Like the diphtheria toxin, the cholera toxin is composed of two types of subunits. Each toxin molecule contains five B subunits that encircle a single A subunit. The B subunit is responsible for binding to the receptor on the mucosa. The A subunit is responsible for activation of adenylate cyclase located on the basolateral cellular membrane (see earlier). A second 10 to 30 kDa LT, zonula occludens toxin, has been described that alters intestinal permeability by acting on intestinal epithelial cell tight junctions.59
Epidemiology
For many centuries, the Bay of Bengal had been considered the “cradle of cholera.” Western countries were relatively free of cholera epidemics until the 19th century, but since then, with the worldwide spread of the disease, six pandemics (across continents) have been reported. We are currently in the seventh pandemic, which started in 1961 in Indonesia and then made its way to the Philippines, Hong Kong, Japan, Korea, Thailand, India, Pakistan, and the Middle East, passing across the African continent to engulf the entire region, and, in 1991, spreading to South America. Although the overall number of cases of cholera in Latin America has subsided since 1991, outbreaks of V. cholerae have continued to occur sporadically throughout sub-Saharan Africa. During 1999, more than 200,000 cases of cholera were reported from Africa, accounting for 81% of the global total of cholera cases.60
Cholera occurs sporadically along the Gulf Coast of the United States, primarily in Texas and Louisiana.61 Among the millions of American travelers to endemic areas in foreign countries, only 41 imported cases of cholera were reported in the United States from 1961 to 1990, and none was associated with secondary spread. The epidemic in South America resulted in 151 cases of cholera in the United States: 26 cases in 1991, 103 in 1992, and 22 in 1993; only one death was reported.62
The South American epidemic that began in Peru in January 1991 reached more than one million cases in its first three years. From 15,000 to 20,000 cases of cholera were reported each week during the peak of the epidemic, for a national incidence of 1 : 1000 persons. Unboiled drinking water, unwashed fruits or vegetables, and food or water from street vendors were implicated risk factors in this explosive outbreak.63,64
The epidemic of V. cholerae O139 Bengal that began in southern India and Bangladesh in late 1992 affected adults predominantly.56 The clinical features of infection with the O139 Bengal strain were virtually indistinguishable from infection caused by V. cholerae O1.65,66
Pathogenesis
Attachment of V. cholerae to the intestinal mucosa is mediated by various surface components, including a fimbrial colonization factor known as toxin-coregulated pilus. The toxin-coregulated pilus attachment protein might play an important role in producing naturally occurring protective antibodies against V. cholerae.67
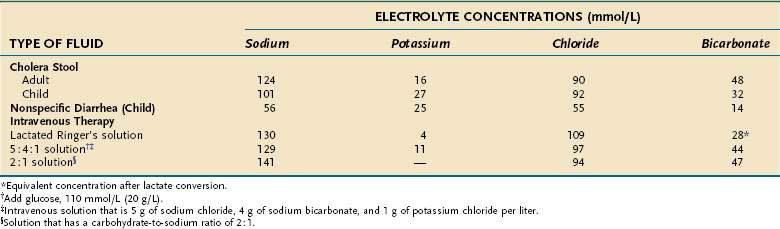
Despite the derivation of the term cholera (Greek: chole, bile), the appearance of choleric stools resembles rice water; that is, the stool has lost all pigment and becomes a clear fluid with small flecks of mucus. The electrolyte composition (Table 107-5) is isotonic with plasma, and the effluent has a low protein concentration. On microscopic examination there are no inflammatory cells, only small numbers of shed mucosal cells.
Table 107-5 Electrolyte Concentrations of Choleric and Nonspecific Fecal Fluid and of Intravenous Fluids Used to Treat Infectious Diarrheas
Clinical Features
Like many other infectious diseases, there is a spectrum of clinical manifestations with V. cholerae, from an asymptomatic carrier state to a desperately ill patient with severe dehydration. The initial stage is characterized by vomiting and abdominal distention and is followed rapidly by diarrhea that accelerates over the next few hours to frequent large volumes of rice-water stools. All the clinical symptoms and signs can be ascribed to fluid and electrolyte losses. Patients present with profound dehydration and hypovolemic shock, usually leading to kidney failure. The stool is isotonic with plasma, although there is an inordinate loss of potassium and bicarbonate, with resultant hypokalemic acidosis (see Table 107-5). Mild fever may be present, but there are no signs of sepsis.
Immunologic Responses
The susceptibility of adults in areas endemic for the O139 Bengal strain of cholera indicates that the afflicted populations are immunologically naive and that prior exposure to V. cholerae O1 does not provide cross-protective immunity. Nevertheless, volunteer challenge studies indicate that an initial infection with O139 Bengal provides protection against recurrent disease.65
Treatment
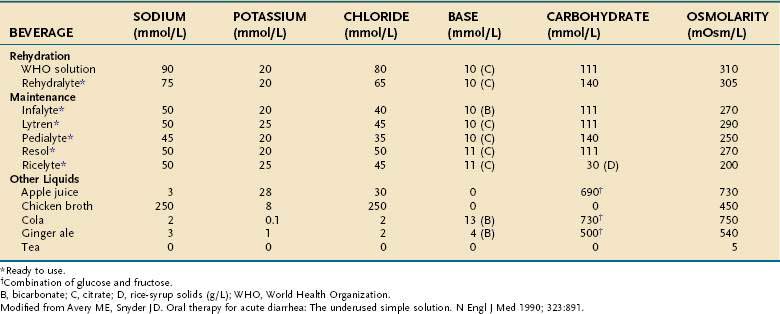
Treatment of acute cholera is based on the physiologic principles of restoring fluid and electrolyte balance and maintaining intravascular volume. These objectives can be accomplished with intravenous solutions or oral fluids that contain electrolytes in isotonic concentrations (see Table 107-5). Particular attention is paid to administration of bicarbonate and potassium, which are lost excessively in choleric stool. Various oral rehydration solutions (ORSs) have been developed for treating mild to moderate cases; ORS is especially useful in developing countries (Table 107-6).68
Antimicrobial agents are useful ancillary measures to treat cholera because their use leads to reductions in stool output, duration of diarrhea, fluid requirements, and Vibrio excretion.69 Tetracycline is recommended at a dose of 40 mg/kg per day orally up to a maximum of 4 g/day in four divided doses for two days; there is no proven value in lengthening the duration of treatment to four days. Single-dose therapy with ciprofloxacin results in a successful clinical response in 94% of patients infected with V. cholerae.70,71 As a result of rising rates of resistance, tetracycline and doxycycline often are less effective than the fluoroquinolones.71,72 Alternative drugs include trimethoprim-sulfamethoxazole (TMP-SMX) and furazolidone.








