Immunodeficiency Diseases
Patients with immunodeficiency syndromes often present with chronic diarrhea, malabsorption, and other intestinal abnormalities, as well as with the effects of the immunodeficiency,
particularly infections. The immunodeficiency syndromes fall into primary and secondary (or acquired) forms (Table 6.45). Primary immunodeficiencies fall into several categories: Those primarily associated with failure to produce antibodies (Table 6.46), those associated with lymphocyte abnormalities, or those involving neutrophils.
particularly infections. The immunodeficiency syndromes fall into primary and secondary (or acquired) forms (Table 6.45). Primary immunodeficiencies fall into several categories: Those primarily associated with failure to produce antibodies (Table 6.46), those associated with lymphocyte abnormalities, or those involving neutrophils.
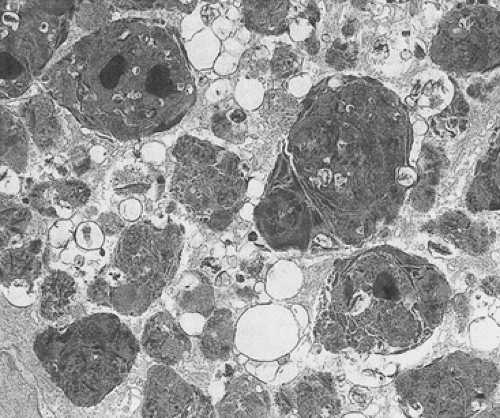 FIG. 6.214. Ultrastructural features of brown bowel disease. Numerous intralysosomal inclusions containing cellular membranes and osmiophilic debris are present. |
TABLE 6.45 Classification of Immunodeficiency Diseases | |
|---|---|
|
Antibody Deficiency Disorders
Selective IgA Deficiency
Selective IgA deficiency is the most common immunodeficiency in Caucasians, being present in about 1 of 600 in the general population as assessed by blood donor screening (588). The incidence varies depending on the population being studied. Published figures range from 1 in 400 in Finland to 1 in 1,500 in Japan (589). IgA deficiency is 10 to 15 times more common in patients with celiac disease than in the general population.
TABLE 6.46 Antibody Deficiency States | |
|---|---|
|
In selective IgA deficiency, IgA, the major mucosal immunoglobulin, is not produced. Patients exhibit an isolated absence or near absence (i.e., <0.10 g/L [<10 mg/dL]) of serum and secretory IgA. In most cases, the cause of the immunodeficiency is unknown. The disease may be congenital or induced by viral infections, leukopenia, and drugs. Unusual cases result from deletion of an IgA gene on chromosome 14 (590). Selective IgA deficiency associates with extended HLA haplotypes that include either a C4A null allele (C4AQ0), 21-hydroxylase gene deletions in the HLA class III region, or rare class IIIC gene haplotypes (591), especially in Caucasians. These haplotypes are rare in Blacks and Asians.
Most patients have defective B-cell maturation with abnormal terminal differentiation of membrane IgA-positive B cells into IgA-secreting plasma cells. A smaller percentage of individuals have a defect in immune regulation of a putative suppressor T cell that selectively inhibits IgA production (592).
Many patients lack clinical abnormalities due to a compensatory increase in IgM. Other patients, especially if deficient in IgG subclasses, present with sinopulmonary disease, diarrhea, malabsorption, autoimmune disease, or bacterial infections, including GI infections (593). Patients with IgA deficiency also frequently have antibodies directed against cow’s milk and ruminant serum proteins, immunoglobulins, thyroglobulin, and collagen. Diseases associated with IgA deficiency are listed in Table 6.47.
Biopsies in IgA-deficient patients may appear completely normal with a seemingly full complement of lymphocytes and plasma cells in the lamina propria, especially in adults. Immunohistochemical analysis for immunoglobulins demonstrates that the lamina propria plasma cells produce IgM and IgG but not IgA. Patients may also have evidence of coexisting celiac disease or bacterial infections (Fig. 6.215). Rarely, patients with a selective IgA deficiency will present with a completely flat mucosa (Fig. 6.216) in the
absence of bacterial overgrowth or Giardia. Such patients may die from severe malabsorption (594).
absence of bacterial overgrowth or Giardia. Such patients may die from severe malabsorption (594).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


