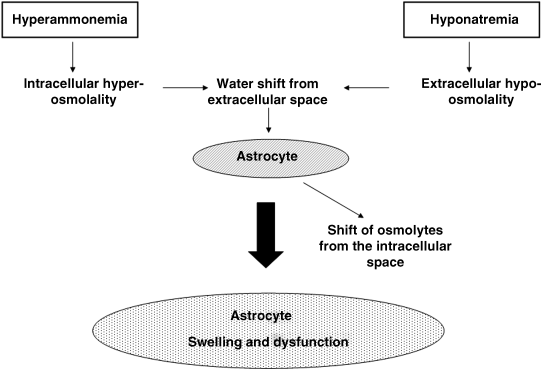
Chapter 18 Salvatore Piano1, Filippo Morando1, and Paolo Angeli1,2 1Department of Medicine (DIMED), University of Padova, Padova, Italy 2Department of Medicine (DIMED), Unit of Hepatic Emergencies and Liver Transplantation, University of Padova, Padova, Italy Unlike hypernatremia, hyponatremia is very common in patients with cirrhosis and ascites. Its prevalence depends on the criteria used to define it. In a prospective study in patients with cirrhosis and ascites, the prevalence of hyponatremia, defined by a serum sodium concentration ≤135 mmol/L, was found to be 28%. Patients with cirrhosis develop two types of hyponatremia: (i) hypovolemic hyponatremia, or (ii) hypervolemic hyponatremia. Hypovolemic hyponatremia is characterized by low serum sodium associated with contraction of plasma volume, lack of edema, ascites, and prerenal renal failure. Hypervolemic or dilutional hyponatremia is due to a marked impairment of renal solute-free water excretion, resulting in a disproportionate renal retention of water with respect to sodium. As a consequence, the most stricking difference between these two conditions lies in the volume status which is reduced in hypovolemic hyponatremia and increased in hypervolemic hyponatremia. Hypervolemic hyponatremia is much more common than hypovolemic hyponatremia in patients with cirrhosis and ascites, thus, this chapter discusses to hypervolemic hyponatremia. Clinically significant hyponatremia (serum sodium value ≤130 mmol/L) in patients with cirrhosis is associated with severe liver disease as reflected by a high Child–Pugh score, poor control of ascites, and serum creatinine value >108 μmol/L [1]. Its pathogenesis is related to a higher rate of renal retention of water in relation to sodium because of a reduction in solute-free water clearance [2–4]. The pathogenesis of water retention in cirrhosis is complex and involves several factors: increased plasma levels of vasopressin (AVP), reduced renal synthesis of prostaglandins, and reduced delivery of filtrate to the ascending limb of the loop of Henle, the diluting segment of the nephron. Patients with cirrhosis and ascites have increased plasma concentrations of AVP which are closely correlated with the reduction in solute-free water excretion [5–7]. Several studies have provided evidence that increased release [2–4] rather than reduced metabolism of AVP [8] is responsible for the increased plasma concentration of AVP in these patients. Taking into account that most patients with cirrhosis and ascites have a degree of hyponatremia and hypo-osmolality that would suppress AVP release in normal subjects [9], a nonosmotic stimulation of AVP release due to a reduction in effective circulating volume is considered the afferent factor leading to increased plasma concentration of AVP. In this context, it should be highlighted that AVP is part of the neurohumoral response to splanchnic arterial vasodilatation which also includes hyperactivation of antinatiuretic and vasopressor systems such as the renin-angiotensin-aldosterone axis and the sympathetic nervous system [9,10]. Bichet et al. [7] showed that the expansion of central blood volume by means of head-out water immersion suppressed plasma renin activity, and the plasma levels of aldosterone, norephineprine, and AVP, resulting in an improvement in both sodium and water excretion. These results were confirmed, almost 20 years later, by clinical studies that showed that in patients with hepatorenal syndrome (HRS) who responded to the prolonged administration of vasoconstrictors and albumin there was a significant improvement in plasma levels of sodium associated with a decrease in the plasma level of AVP [11,12]. The first step in the action of AVP on water excretion is its binding to V2 receptors on the basolateral membrane of the principal cells of the collecting duct. The activation of the V2 receptors stimulates adenyl cyclase via the stimulatory G protein and promotes the formation of cyclic adenosine monophosphate (cAMP). The post-cAMP events that mediate the increase in water permeability are related to the presence of a number of the water channel family that are exclusively expressed in the principal cells of the collecting duct, aquaporin-2. AVP stimulates the translocation of aquaporin-2 from sub-apical multivescicular bodies to the apical membrane [13]. According to this pathogenetic interpretation of hyponatremia in cirrhosis, urinary excretion of aquaporin-2 would be expected to be increased and not reduced as was found by Esteva-Font et al. [14]. The authors found urinary aquaporin-2 excretion in patients with cirrhosis and hyponatremia lower than in patients without hyponatremia and did not find a correlation between plasma level of AVP and urinary aquaporin-2 excretion. Nevertheless, the reduced urinary excretion of aquaporin-2 in patients with cirrhosis and hyponatremia may represent the molecular basis of the phenomenon of escaping the effects of AVP similar to that found in healthy subjects [14]. In fact, if the AVP–aquaporin-2 system was persistently activated in cirrhosis, solute-free water reabsorption would be permanently increased relative to sodium retention, and serum sodium levels would decline steadily to levels incompatible with life. Fortunately, this does not happen in clinical practice as, in most patients with cirrhosis, hyponatremia, when developed, is stable for weeks or even for a few months. Therefore, in patients with cirrhosis who develop hyponatremia, a reduced availability of aquaporin-2 may be part of a protective mechanism that counteracts the AVP-induced increase in solute-free water reabsorption in the collecting ducts and the further fall in serum sodium concentration. Two additional factors are thought to be relevant in the pathogenesis of hyponatremia in cirrhosis: a reduced production of solute-free water due to reduced sodium delivery to the distal tubule and increased release of renal prostaglandins [15]. The former may be the consequence of a reduction in glomerular filtration rate (GFR) or an increase in sodium reabsorption in the proximal tubule [16,17]. As a consequence of its complex pathogenic mechanisms, impaired water excretion in cirrhotic patients with ascites may be induced and/or worsened by several factors including diuretics, nonsteroidal anti-inflammatory drugs (NSAIDs) and other drugs [18–20], as well as by large-volume paracentesis if not followed by adequate plasma expansion [21], and bacterial infections. As far as diuretics are concerned, it should be stated that they can precipitate hypovolemic hyponatremia causing an excessive increase in sodium excretion and thus dehydration, as well as hypervolemic hyponatremia. With reference to the latter, it should be noted that loop diuretics may directly interfere with the urine diluting process by impairing free water generation through the inhibition of NaCl reabsorption in the thick ascending limb of the loop of Henle. On the other hand, any type of diuretic may stimulate a further release of AVP by means of a further reduction in the effective circulating volume [22]. Finally, among the drugs that can precipitate hyponatremia in cirrhosis, an acute reduction in serum sodium concentration is common during treatment with terlipressin, an AVP agonist, for severe portal-hypertensive bleeding. It develops rapidly after the start of therapy, may be severe in some patients, is associated with neurologic complications, and is usually reversible after terlipressin withdrawal [23]. The symptoms of hyponatremia (anorexia, apathy, nausea, vomiting) associated with cirrhosis and ascites do not differ from those of hyponatremia in other diseases in which hyponatremia is coupled with an increase in extracellular fluid volume. Few data exist on the clinical consequences of hyponatremia in patients with cirrhosis because hyponatremia almost always occurs in the setting of advanced liver failure, which causes a wide array of clinical manifestations. Nevertheless, from a clinical point of view, hyponatremia was found to be associated with a greater likelihood of hepatic encephalopathy, hepatorenal syndrome (HRS), and spontaneous bacterial peritonitis (SBP) [1]. The relationship between hyponatremia and HRS and/or SBP should probably be viewed from a pathophysiologic perspective. HRS and SBP can precipitate hyponatremia in patients with cirrhosis [24] because they are characterized by a further impairment of the effective circulating volume and, thus, to a maximal activation of all the factors that are involved in the physiopathology of hyponatremia. Nevertheless, it is also proven that hyponatremia represent a significant risk factor for the development of HRS in patients with cirrhosis [25]. Both these clinical conditions further impair the cardiocirculatory system and further increase the plasma levels of AVP, thus favoring the development of hyponatremia in patients with cirrhosis and ascites. However, the terms of the relationship must be reversed when considering encephalopathy [1], because hyponatremia appears to precipitate and aggravate hepatic encephalopathy in patients with cirrhosis. In our experience, serum levels of sodium and ammonia appear to be the major determinants of electroencephalographic abnormalities in patients with cirrhosis [26]. In addition, the existence of hyponatremia was found to be a major risk factor for the development of overt encephalopathy in all patients with cirrhosis and ascites [27], particularly in those with refractory ascites [28]. Finally, it has been observed that in patients with cirrhosis who were treated with transjugular intrahepatic portosystemic shunt, hyponatremia was a major risk factor for hepatic encephalopathy [29]. In order to explain these clinical observations it may be hypothesized, on the basis of experimental and human studies [30–33], that an increased ammonia level and reduced sodium concentration act synergistically to induce swelling of astrocytes (Figure 18.1). The consequent cerebral edema would not be sufficient to cause an increase in intracranial pressure, but astrocyte swelling would result in a number of alterations in neurologic function, which would facilitate the development of hepatic encephalopathy. Other neurologic manifestations such as headache, focal motor deficits, seizures, and cerebral herniation are very uncommon in patients with cirrhosis compared with other patients who develop hyponatremia. As it has been stated that the most important factor in determining the severity of neurologic symptoms is the rate of fall in serum sodium concentration, it is likely that the relatively low incidence of neurologic manifestations in patients with cirrhosis and hypervolemic hyponatremia is related to the fact that in most of these patients hyponatremia is chronic rather than acute, and this gives sufficient time for the brain to adjust to the reduced osmolality of the extracellular fluid. Figure 18.1 Pathogenesis of hepatic encephalopathy: role of hyponatremia. For several years hyponatremia has been recognized as a powerful prognostic indicator in patients with cirrhosis and ascites [34]. The presence of hyponatremia is associated with a low probability of survival in patients with cirrhosis and ascites [35] as well as in patients with acute-on-chronic liver failure [36]. In addition, it has been shown that hyponatremia is an independent predictive factor of the impaired health-related quality of life in these patients [35]. Nevertheless, full recognition of the prognostic value of reduced serum sodium concentration in patients with advanced cirrhosis has occurred with the development of the Model of end-stage liver disease (MELD) as reliable indicator of 90-day survival in patients who are candidates for liver transplantation (LT). This scoring system utilizes three widely available laboratory values: total bilirubin (mg/dL), creatinine (mg/dL), and international normalized ratio (INR) of the prothrombin time [37,38]. Creatinine was initially thought to capture the prognostic implications of all the clinical complications associated with the circulatory derangements in cirrhosis: ascites, hyponatremia, and renal failure. However, it has been subsequently reported that hyponatremia, uncontrolled ascites [39], as well as type 1 HRS were predictive of short-term mortality independently of the MELD score [40]. From a speculative point of view it is relevant to note that the independent predictive value of serum sodium concentration disappears when GFR measured by means of insulin clearance was incorporated into the MELD score [41]. The independent prognostic significance of low serum sodium concentration was subsequently confirmed by several authors worldwide and a variety of prognostic models incorporating serum sodium and MELD have been proposed [42–44]. The most advanced is that of Kim et al. [45] which is based on the very simple concept that “the lower MELD the higher prognostic relevance of hyponatremia”. Patients with cirrhosis and hyponatremia are at increased risk of developing neurologic complications including central pontine myelinolysis after LT [46,47]. Although central pontine myelinolysis is thought to be brought about by a rapid change in serum sodium in the early postoperative period rather than by hyponatremia per se, it can be considered as a hyponatremia-related complication. Two recent studies have shown that the existence of hyponatremia before LT in patients with cirrhosis is associated not only with an increased risk of neurologic complications after LT, but also with an increased risk of renal failure and infectious complications, longer duration of hospital stay, and, more importantly, increased short-term mortality after LT [46,47]. More recently, however, the effect of pretransplant hyponatremia on short-term mortality after LT has not been confirmed [48]. The first step in the management of hyponatremia in cirrhosis is to identify whether hyponatremia is hypovolemic or hypervolemic, because management differs markedly. The management of hypovolemic hyponatremia is essentially based upon the administration of sodium with the aim of normalizing the depleted body sodium stores, while the key to management of hypervolemic hyponatremia is to increase renal solute-free water excretion. The administration of hypertonic sodium chloride cannot be recommended, on a regular basis, for the management of hypervolemic hyponatremia because it has a partial and short-lived effect in increasing serum sodium concentration in cirrhosis and it further increases ascites and edema in these patients. The treatment of hypervolemic hyponatremia has some ambitious aims. First, as hyponatremia has been shown to be a predisposing factor for hepatic encephalopathy in some studies [27,28], the improvement of serum sodium concentration may help to reduce the risk of this complication. Second, treatment of hyponatremia may improve survival and quality of life in patients with cirrhosis [35]. Third, in patients awaiting LT, the normalization of serum sodium concentration before transplantation may help to reduce the frequency and severity of neurologic complications after LT [46,47]. Unfortunately, because of the lack of pharmacologic agents, these potentially beneficial effects of treating hyponatremia in cirrhosis have not been evaluated in prospective randomized controlled trials (RCTs). The traditional options for treatment of hypervolemic hyponatremia in cirrhosis include: (i) fluid restriction with or without reduction of the doses/withdrawal of diuretics, and (ii) the administration of albumin. Fluid restriction alone or associated with a reduction of the doses/withdrawal of diuretics, in particular of loop diuretics, has been the standard of care for the management of hypervolemic hyponatremia in cirrhosis for many years. Nevertheless, its efficacy in improving serum sodium is modest and it is probably only helpful in preventing a further reduction in serum sodium concentration. In some prospective randomized studies comparing the efficacy of vaptans with placebo, in which patients from both groups were treated with fluid restriction, the efficacy of this intervention in improving serum sodium concentration (>5 mmol/L) ranged from 0% to less than 30% [49–51]. Some reports in a small series of patients and the preliminary results of a still unpublished RCT suggest that the administration of albumin (20–40 g/day) for a short period of time improves serum sodium concentration in patients with hypervolemic hyponatremia [52–54]. This beneficial effect of albumin is probably related to an improvement in the effective circulating volume with a consequent reduction in the activity of the sodium and water-retaining systems, including AVP. A new family of drugs, known as vaptans (Table 18.1), which increase renal solute-free water excretion through the inhibition of the V2 receptors located in the principal cells of the collecting ducts, has recently been investigated in the management of hypervolemic hyponatremia associated with different conditions including cirrhosis [55–57]. Nevertheless, among the different compounds, only tolvaptan has been approved, limited to Japan and the United States, for the management of hyponatremia in cirrhosis. RCTs have demonstrated that the vaptans, including lixivaptan, tolvaptan, and satavaptan, are effective in improving serum sodium concentration in patients with hyponatremia [58–61]. In short-term studies, normalization of serum sodium concentration was observed in 27–54% of patients. In particular, the rate of normalization of serum sodium concentration in patients with cirrhosis was 33% by week 4 [59,62]. The most common side effect of the vaptans is thirst, which is related to the pharmacodynamic effect of these drugs. The long-term effects of tolvaptan on serum sodium concentration was evaluated in patients with cirrhosis, in patients with chronic heart failure (CHF), and in those with syndrome of inappropriate secretion of AVP (SIADH). More than 60% and 45% of patients in the mild and marked hyponatremia groups, respectively, exhibited normal serum sodium levels by week 4. Correction rates seemed to be generally similar among patients with CHF and SIADH/other but may have been somewhat lower among patients with cirrhosis (Figure 18.2). Mean dosage of tolvaptan was 30 mg/day [59]. Hyponatremia recurred in tolvaptan-treated patients after discontinuation [62]. The long-term effect of satavaptan has been evaluated in three randomized double-blind studies comparing satavaptan with placebo in patients with cirrhosis and uncomplicated ascites as well as in those with difficult-to-treat ascites. Satavaptan was more effective than placebo in improving the serum sodium concentration in patients with hyponatremia. Satavaptan was not more effective than placebo in the control of ascites in any of the populations studied but, overall, in one of the studies mortality was higher in patients treated with satavaptan than with placebo [63]. Table 18.1 Vaptans. Figure 18.2 Rate of normalization of serum sodium concentration during long-term treatment with tolvaptan (adapted from ref. no 59).
Hyponatremia and Other Electrolyte/Ion Disorders
Hyponatremia
Epidemiology and Pathogenesis
Clinical Consequences and Prognostic Implications
Management
Compound
Conivaptan
Lixivaptan
Satavaptan
Tolvaptan
Phase
2
2
3
Approved by FDA
Receptor
V1a/V2
V2
V2
V2
Route of administration
i.v.
oral
oral
oral
Urine volume
↑
↑
↑
↑
Urine osmolality
↓
↓
↓
↓
Sodium excretion/24 hours
↔
↔*
↔*
↔
Company
Astellas Pharma
Cardiokine
SanofiAventis
Otsuka
*= increase 24 hour sodium excretion at high dose. 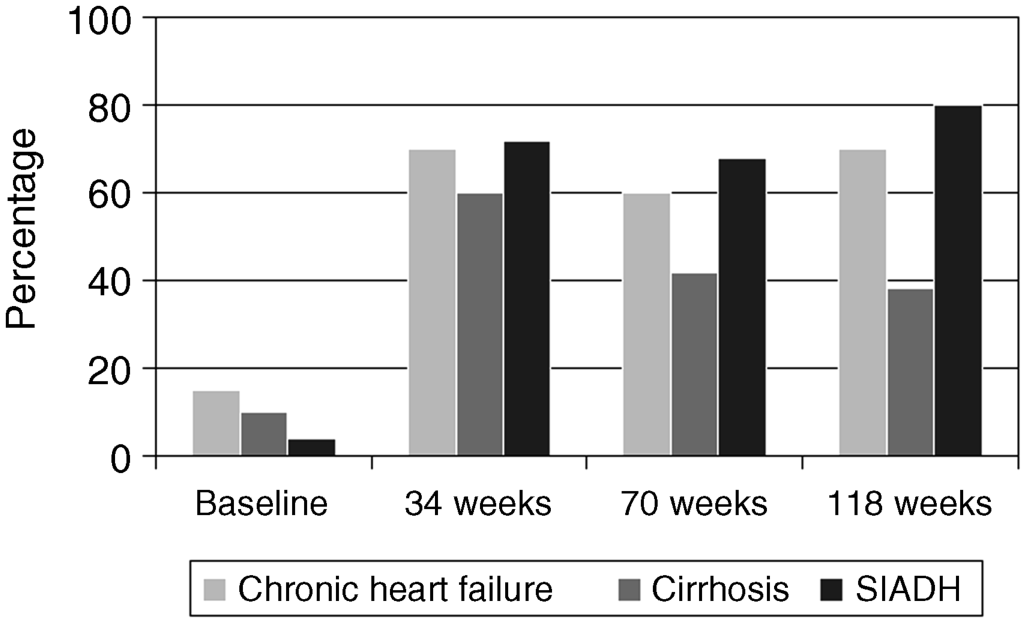
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


