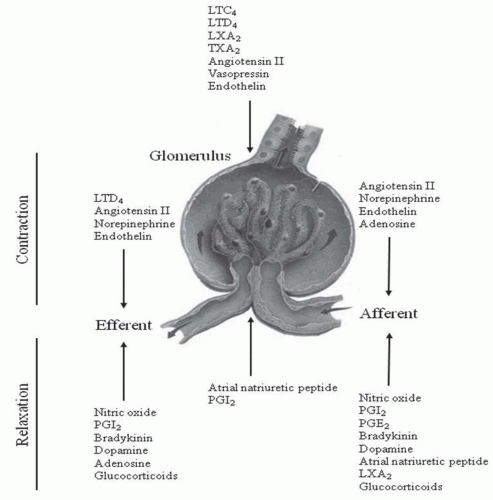
 FIGURE 8.1 Hormones regulating glomerular, afferent, and efferent arteriolar contractility. Glucocorticoid action may be pharmacologic and indirect. LTC4, leukotriene C4; LTD4, leukotriene D4; LXA2, leukotriene A2; TXA2, thromboxane A2; PGE2, prostaglandin E2; PGI2, prostacyclin. |
nucleus solitarius in the brainstem, from which postsynaptic pathways project to the magnocellular neurons. Whereas a 5% to 8% decrease in blood volume or systemic arterial pressure has little effect, further hemodynamic compromise leads to a steep increase in circulating AVP levels. Significant reductions (10%-30%) in circulatory arterial volume or blood pressure can override osmoregulation and result in markedly increased AVP levels in the face of decreased plasma osmolality.19 Other less potent stimuli for AVP secretion include fever, emesis,20 and oropharyngeal osmoreceptors.21
TABLE 8.1 Hormonal Modulation of Tubular Transport | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
initiating the second-messenger cascade responsible for the cellular actions of AVP.25 Stimulation of V1R, although not directly involved in control of tubular water and electrolyte transport, increases sodium excretion because of the influences on blood pressure, effective arterial circulating volume, glomerular filtration rate, and circulation in the vasa recta system.27,28 Additional biologic effects of AVP mediated through V1Rs include platelet aggregation29 and increased glycogenolysis and gluconeogenesis in the liver.30
TABLE 8.2 Vasopressin Receptor Types, Genetics, Location, and Main Physiologic Effects | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
which is necessary for ENaC activity. Finally, V2Rs activate urea transporters, such as UTA1, in the distal nephron.43,44,45 This increase in urea reabsorption and recycling maximizes sodium reabsorption in the TAL by supporting the axial hyperosmotic gradient drawing water from the distal nephron.46 The clinical importance of the V2R in water balance disorders is underlined by the current use of V2R antagonists in a clinical setting (see later).
(NSIADH) may be responsible for this picture. In fact, in some children, it appears to be due to an activating mutation of V2R. In other patients, it may be due to abnormal control of aquaporin-2 water channels in renal collecting tubules or production of an antidiuretic principle other than AVP.69 In a study with SIADH patients, treatment with V2R antagonists, commonly referred to as vaptans, increased serum Na+ concentration and decreased its excretion. Tolvaptan has been recently approved in the United States and Europe for the treatment of hyponatremia associated with SIADH, as well as cirrhosis and congestive heart failure. Recently, a dual vasopressin V1R and V2R antagonist, conivaptan, improves hyponatremia in rats with SIADH, suggesting a therapeutic potential for conivaptan in the treatment of SIADH.70
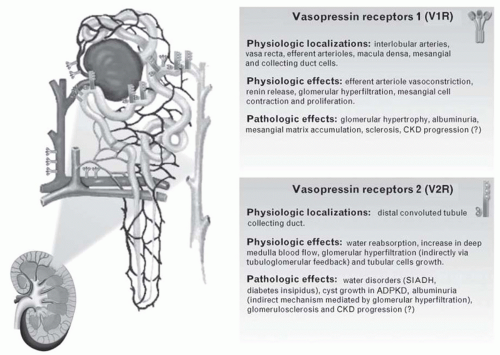 FIGURE 8.2 Vasopressin receptors: physiology and pathologic involvement in renal diseases. (Reproduced from Bolignano D, Zoccali C. Vasopressin beyond water: Implications for renal diseases. Curr Opin Nephrol Hypertens. 2010;19(5):499-504.) |
in the PREVEND study.79 However, V2 receptors have not been found in glomeruli or proximal tubules, suggesting indirect effects. Recent evidence suggests a strong interaction between AVP and the renin-angiotensin system (RAS).80,81 In fact, chronic RAS blockade by angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) prevents the desmopressin-induced albuminuria, indicating that RAS mediates the effects of AVP on glomerular hemodynamics.82 Recent studies using V1R knockout mice show that AVP regulates body fluid homeostasis and the GFR by activating RAS through V1Rs in MD cells, and subsequently the V2R-aquaporin 2 system.83 Simultaneous AVP and RAS blockade may represent a good therapeutic approach for delaying renal disease progression.
concentrations of angiotensinogen in the proximal tubule of anesthetized rats greatly exceeded the free angiotensin I and II concentrations110; and fourth, human angiotensinogen was not detected in urine of normotensive rats infused with the molecule.111
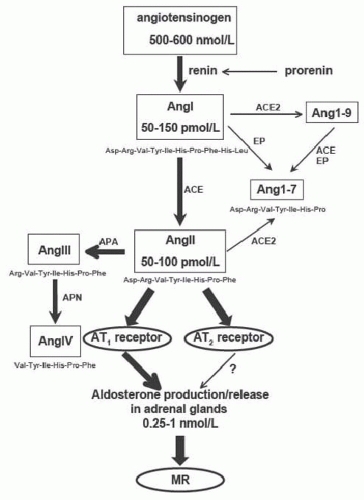 FIGURE 8.3 Schematic representation of the renin-angiotensin system showing plasma concentrations of some components as measured in anesthetized rats. EP, endopeptidase; APA, aminopeptidase A; APN, aminopeptidase N. |
dogs136 and its inhibition attenuates the pressor response to norepinephrine in humans.137 Experimental data suggest the presence of a local renin-angiotensin system in the vasculature that contributes to the regulation of vascular tone.112
direct stimulation of Na/Pi cotransport activity as a result of increase in the expression of brush-border membrane NaPi-IIa protein level and that stimulation is most likely mediated by posttranscriptional mechanisms.164
system is, indeed, functional.156 Some investigators have proposed that this local system plays a role in proximal tubule NaCl and HCO3 absorption, pathogenesis of essential hypertension, and expression of the phenotype of autosomal dominant polycystic kidney disease.156 Independent regulation of renal Ang II production has not been definitively demonstrated. Circulating Ang II stimulates renal angiotensinogen mRNA production and intact urine angiotensinogen suggests its presence along the whole nephron and that renin and ACE activity are available all through the nephron.126
TABLE 8.3 Conditions Associated with Increased Levels of Circulating Atrial Natriuretic Peptide | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
through apical sodium channels.289 It appears that the natriuretic effect of RNP is more potent than that of ANP, possibly because it is resistant to degradation by renal cortical metalloendopeptidase.290 Systemic infusion of RNP results in effects similar to those of ANP99-126.
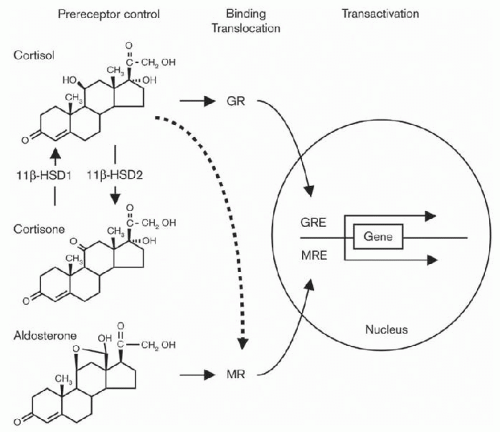 FIGURE 8.4 The intracellular concentrations of steroid molecules available for binding to glucocorticoid receptor (GR) or mineralocorticoid receptor (MR) depend on the free extracellular concentrations available for diffusion into the cytoplasm and the intracellular prereceptor control mechanism constituted by the 11β-hydroxysteroid dehydrogenase type 1 and 2 (11β-HSD1, 11β-HSD2) enzymes. Whereas 11β- HSD1 acts predominantly as a reductase and converts the 11-ketosteroid cortisone with virtually no affinity for MR and GR into the 11β-hydroxyglucocorticoid cortisol with a high affinity for both GR29,58 and MR, 11β-HSD220 is exclusively an oxidase and inactivates cortisol into cortisone, which allows protection of MR-expressing cells from promiscuous activation of MR by the glucocorticoid hormone cortisol. GRE, glucocorticoid-response element; MRE, mineralocorticoid-response element. (Frey FJ, Odermatt A, Frey BM. Glucocorticoid-mediated mineralocorticoid receptor activation and hypertension. Curr Opin Nephrol Hypertens. 2004;13:451-458.) |
sodium reabsorption320 or early stimulation of salt intake,321 possibly via phospholipase C/PKC signaling pathways.
potassium excretion, so prolonged use of such agents may enhance potassium retention and predispose a patient to aldosterone breakthrough. Recent studies in rat models of metabolic syndrome with early nephropathy have shown enhanced aldosterone secretion due to adipocyte activity that was not abolished by candesartan administration.35 These results suggest that adipocyte-released factors outside of Ang II may enhance aldosterone secretion and lead to increased proteinuria and podocyte injury in rats.35 Thus, potassium, angiotensin II, and adipocyte-released factors may all contribute to the increase in aldosterone secretion in patients on prolonged ACE inhibitors or ARB therapy. The exact definition of aldosterone breakthrough has been a subject of controversy, as there is no current consensus on its precise definition. One of the common definitions is a rise in plasma aldosterone concentration, often past baseline values, following an initial decrease after the initiation of ACE inhibitor or ARB therapy.322
treatment of renal tubular epithelial cells increases calcium inflow and intracellular cyclic adenosine monophosphate levels. The results suggest that aldosterone plays a pivotal role in tubulointerstitial fibrosis by promoting tubular epithelial-mesenchymal transition and collagen synthesis in proximal tubular cells. The process is MR-dependent, and mediated by ERK1/2 mitogen-activated protein kinase pathway.346
permeability of the collecting tubule.360 In addition, adrenal corticosteroids contribute to urinary concentration by stimulating Na, K, and HCO3 transport in the thick ascending limb of Henle’s loop.361 It is not entirely clear whether glucocorticoids, mineralocorticoids, or both mediate the effects of corticosteroids on renal-concentrating mechanisms.
increase Cl–-HCO3– exchange and H+-K+-ATPase activity in the collecting duct.371 The latter effect results in enhanced K+ reabsorption by type A intercalated cells (and an apparent decrease in K+ secretion).371 A potential mechanism of action involves β-adrenergic stimulation of cAMP production and subsequent conversion to adenosine.381
major actions of this system are mediated by bradykinin, a peptide hormone that exerts potent proinflammatory and vasodilatory effects. Interestingly, all components of the KKS are also expressed in the kidney, especially in the distal convoluted and connecting tubule as well as in the collecting duct, and have been shown to regulate renal hemodynamic and tubular function. In addition, in the last few years, studies have linked the KKS to different pathologic states including the diabetic nephropathy as reviewed here.
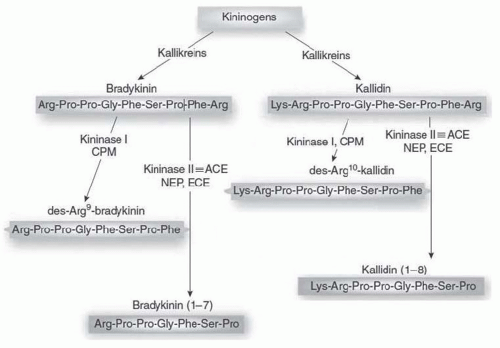 FIGURE 8.5 Biosynthesis and metabolism of kinins. CPM, carboxypeptidase-M; ACE, angiotensin I-converting enzyme; NEP, neprilysin (endopeptidase 24.11); ECE, endothelin-converting enzyme; red, active peptides; blue, inactive peptides. (Adapted from Kakoki M, Smithies O. The kallikrein-kinin system in health and in diseases of the kidney. Kidney Int. 2009;75(10):1019-1030.) (See Color Plate.) |
involving membrane depolarization of kallikrein-secreting cells in the renal connecting tubules, followed by enhanced calcium influx.406,407 In addition to dietary sodium and potassium intake, hereditary factors may determine tissue kallikrein activity. In fact, a loss of function polymorphism in the human tissue kallikrein gene (R53H) has been identified with a frequency of 0.03 in Caucasians.408 Interestingly, these partially tissue kallikrein-deficient subjects develop a form of arterial dysfunction characterized by remodeling of the brachial artery, which is not adapted to a chronic increase in wall shear stress.408
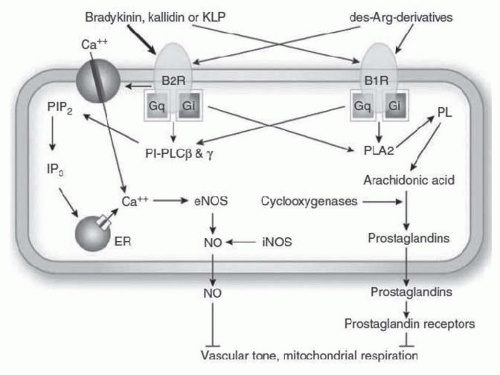 FIGURE 8.6 Bradykinin intracellular signaling cascade. The thickness of arrows arising from the kinins indicates the relative potency of each peptide to elevate intracellular calcium concentrations. PIP2, phosphatidylinositol-4,5-bisphosphate; PI-PLC, phosphatidylinositolspecific phospholipase C; IP3, 1,4,5-inositol triphosphate; ER, endoplasmic reticulum; PL, phospholipids; PLA2, phospholipase A2. (Reproduced from Kakoki M, Smithies O. The kallikrein-kinin system in health and in diseases of the kidney. Kidney Int. 2009;75(10):1019-1030.) |
kallikrein.405
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


