An endocrinologic evaluation of patients who have male-factor infertility has clearly evolved and leads to specific diagnoses and treatment strategies in a large population of infertile men. A well-considered endocrine evaluation is especially essential with the ever-growing popularity of assisted reproductive techniques and continued refinements with intracytoplasmic sperm injection.
In addition to the surgical correction of a varicocele, the evaluation and management of the various endocrinopathies encountered in urologic practices remains invaluable in the management of the infertile male. The question posed at the time of this publication is “has it evolved?” In their retrospective review of 1035 patients in 1997, Sigman and Jarow found that 9.6% of their patients had abnormal endocrine studies. The most common finding was an isolated serum elevation of follicle-secreting hormone (FSH) in 7.9% of the patients. FSH may be elevated in patients who have abnormal spermatogenesis caused by Sertoli cell dysfunction and inadequate negative feedback inhibition by inhibin. Elevation of FSH above 7.6 mIU/mL along with a testis axis of less than 4.6 cm has a probability of nonobstructive azoospermia (NOA) of 89% .
The present authors believe, however, that the presence of a normal semen analysis does not always guarantee a eugonadal state. Furthermore, it is widely recognized that a normal FSH level does not always guarantee the presence of intact spermatogenesis.
Steroidogenesis
Testosterone is the primary circulating androgen in the human male. Testosterone release from the Leydig cells accounts for approximately 95% of the total testosterone production, with the remaining 5% secreted by the adrenal cortex.
Androgen biosynthesis in the testes starts with the formation of cholesterol. Cholesterol is produced from de novo synthesis from acetate, via low-density lipoprotein delivery and preformed cholesterol esters present in the Leydig cells. Cholesterol then is converted to pregnenolone under the influence of luteinizing hormone (LH). This conversion is the rate-limiting step in the testicular synthesis of testosterone. Testosterone then can be converted further to either dihydrotestosterone (DHT) via 5-alpha-reductase or estradiol via the P-450 aromatase pathways. Aromatase is present in the testes, brain, skin, liver, and adipose tissues . Testosterone and DHT bind to the same androgen receptor. Estradiol binds to its own receptor.
Once one of these androgens binds to the receptor complex, DNA transcription ensues, and several key physiologic and developmental effects are produced. The effect is specific to the target tissue. There are a variety of androgen target tissues. Some respond directly to testosterone and some to DHT ( Box 1 ). Developmentally, testosterone stimulates the Wolffian duct system (epididymis, vas deferens, and seminal vesicles). Testosterone also is converted in the genital folds to DHT, which is very important for genital fold development. This conversion facilitates the closure of the labio-scrotal folds and results in the urethral meatus assuming its distal-most position.
Testosterone mediated
- •
Brain
- •
Breast
- •
External genitalia enlargement
- •
Internal genitalia differentiation
- •
Liver
- •
Muscle
- •
Testis
Dihydrotestosterone mediated
- •
External genitalia development
- •
Hair follicle
- •
Prostate
- •
Sebaceous gland
- •
Seminal vesicles
Both FSH and testosterone are essential for normal functioning of the seminiferous tubules in vitro and in vivo. Testosterone is required for completion of the meiotic division and spermatid development and thus plays an important role in the initiation and maintenance of spermatogenesis . After the third and final rise of testosterone that occurs at puberty, the serum level is maintained near 6 ng/mL (600 ng/dL) during adult life until it begins to decline gradually with aging .
Hypogonadism
By traditional epidemiologic methods, an estimated 1 in 200 men have abnormally low levels of testosterone . Hypogonadism is defined as a low total testosterone level (Food and Drug Administration [FDA] normal range, 300–1000 ng/dL) that can be associated with clinical findings such as decreased libido, infertility, anemia, mood changes, alterations in body hair distribution, and decreases in lean muscle mass and bone mineral density. Although men initially have 700 million Leydig cells, they lose approximately 6 million per year after age 20 years . Nieschlag and colleagues , however, did not find any significant differences in sperm counts or fertilizing capacity between younger men and men beyond their seventh decade. Hypogonadism can be separated further into primary and secondary forms. Primary hypogonadism is defined as a low serum testosterone level with normal or elevated LH. Serum FSH levels may be normal or elevated, as seen in patients who have Klinefelter’s syndrome, anorchia, or other iatrogenic causes.
Hypothalamic-pituitary axis abnormalities
Secondary hypogonadism can be categorized further into congenital forms such as idiopathic hypogonadotropic hypogonadism (IHH) ( Box 2 ) or acquired forms. IHH is defined as a low serum testosterone level associated with a low LH level with or without a low FSH level. An important caveat is that low LH levels do not always correlate with low FSH levels, and visa versa. Acquired forms of inappropriately low LH levels include hypopituitarism, hyperprolactinemia, estradiol excess, and noncongenital idiopathic causes. The clinical presentation and the diagnostic implications are based in part on whether this form of androgen deficiency is pre- or postpubertal.
Congenital idiopathic hypogonadism
- •
Anosmic (Kallman syndrome)
- •
Nonanosmic: normosmic idiopathic hypogonadotropic hypogonadism
- •
Fertile eunuch syndrome
Adrenal hypoplasia congenital
Genetic defects of the gonadotropin subunits
- •
Follicle-stimulating hormone-beta mutations
- •
Luteinizing hormone-beta mutations
- •
Mutations in leptin and leptin receptor genes
- •
Hypogonadotropic hypogonadism associated with other pituitary hormone deficiencies
- •
PROP-1 mutations
- •
HERS1 mutations
- •
Complex syndromes that include hypogonadotropic hypogonadism
- •
Prader-Willi syndrome
- •
Congenital spherocytosis
- •
Moebius syndrome
- •
Cerebellar ataxia
- •
Retinitis pigmentosa
- •
Kallmann syndrome is the anosmic form of IHH. Associated clinical features may include neurologic abnormalities (synkinesia, oculomotor deficits, deafness, mental retardation, and cerebellar defects,) unilateral renal agenesis, midline facial defects, and pes cavus. Kallmann syndrome is a common X-linked recessive form of human hypogonadotropic hypogonadism . In a recent report from Bhagavath and colleagues , KAL1 mutations were an uncommon cause of Kallmann syndrome in male patients, occurring in about 3.7% of all IHH males and in 6.3% of anosmic/hyposmic IHH males in their study population. IHH thus is probably far more common that previously expected, with patients who have Kallmann syndrome representing merely the “tip of the iceberg.”
Pituitary
Hypopituitarism is the partial or complete insufficiency of anterior pituitary hormone secretion and may result from pituitary or hypothalamic disease. The reported incidence (12–42 new cases per million population per year) and prevalence (300–455 per million population) probably are underestimated. Clinical manifestations depend on the extent of hormone deficiency and may be nonspecific (eg, fatigue, hypotension, cold intolerance) or more specific (eg, growth retardation secondary to insufficiency of growth hormone or impotence and infertility as a result of gonadotropin deficiency). A number of inflammatory, granulomatous, or neoplastic diseases as well as traumatic or radiation injuries involving the hypothalamic-pituitary axis can lead to hypopituitarism .
Recent observations
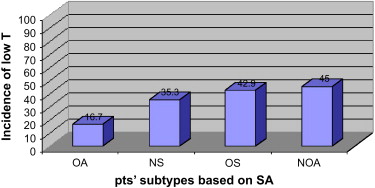
At the University of Illinois in Chicago, the authors recorded the incidence of hypogonadism in various diagnostic categories of patients presenting to an infertility clinic by retrospectively reviewing the charts of 120 consecutive patients. Inclusion criteria were diagnoses of NOA, obstructive azoospermia (OA), oligospermia defined by World Health Organization (fourth edition) criteria, and men who had normal semen analyses. Serum testosterone was measured by electrochemiluminescent immunoassay or turbulent flow liquid chromatography tandem mass spectrometry; FDA criteria defined hypogonadism as a morning serum testosterone level less than 300 ng/dL. Interestingly, the incidence of hypogonadism in men who had NOA was 45.0%. The incidence of hypogonadism was 42.9%, 35.3%, and 16.7%, respectively in men who had oligospermia, normal semen analyses, and OA ( Fig. 1 ). This subset of men who have OA may be considered a negative control because of the relatively exclusive nature of this anatomic condition. The fact that the incidence of hypogonadism in the OA group parallels that of the general population serves to validate the expectation that the OA group serves as a control. The lack of compensatory elevation of LH above 12.0 IU/L in the patient population suggested a form of hypogonadotropic hypogonadism. The finding that one third to nearly one half of the men who had NOA had an incidence of hypogonadism by FDA criteria supports the thesis that hypogonadotropic hypogonadism may be considerably more prevalent in the infertile male population than previously understood.
Hyperprolactinemia
Hyperprolactinemia suppresses both FSH and LH and may be caused by medications, concurrent medical illnesses, stress (both physiologic and psychologic), or pituitary tumors or may be idiopathic. Prolactin is produced in the anterior pituitary; it is responsible for lactation in women but has negligible physiologic effects in men. Normal prolactin levels in men should be less than 18 ng/dL, and testing should be repeated if the level is elevated, because biologic variability is particularly high with this assay. The most common medications that induce hyperprolactinemia are phenothiazines, imipramine, methyldopa, and reserpine. The clinician also must consider the possibility of prolactinomas, both micro- and macroadenomas. Symptoms of prolactinomas include infertility, depressed libido, galactorrhea, headache, fatigue, and erectile dysfunction.
A small percentage of hyperprolactinemic men who have adenomas may have borderline-normal serum testosterone levels . Brain MRI with gadolinium enhancement is diagnostic of anatomic pituitary pathology. Most cases of prolactin-secreting microadenomas respond to medical treatment with either bromocriptine or cabergoline. Both function in a similar way as dopamine agonists. Cabergoline has a longer half-life and therefore offers the advantages of fewer side effects and less frequent dosing. Patients who have idiopathic hyperprolactinemia also may be treated with medication, which may be withdrawn yearly to determine whether hyperprolactinemia persists .
Thyroid status
Thyroid diseases, both hyper- and hypofunction, can have an adverse impact on male reproduction. A lack of consensus exists, mainly because of the paucity of well-controlled studies, on the exact effect of thyroid abnormalities on the male reproductive system. Hyperthyroidism seems to affect both steroid hormone metabolism and sperm quality. Evidence of consistent elevations of sex hormone–binding globulin (SHBG) and total testosterone is presented in the literature; however, serum levels of free testosterone usually are not affected .
More recently, several groups of investigators examined the effects of hyperthyroidism on semen quality. Abalovich and colleagues observed the following alterations in semen analysis in a series of 21 patients who had hyperthyroidism: asthenospermia in 85.7%, hypospermia in 61.9%, oligospermia in 42.9%, necrospermia in 42.9%, and teratospermia in 19.0%. These alterations seem to be reversible, because 85% were normalized when a euthyroid state was re-established. In 2002, Krassas and colleagues , in a prospective, controlled study compared semen parameters in 23 thyrotoxic males and 15 healthy controls. These investigators reported that mean semen volume was normal for both groups, but mean sperm density and motility were lower in the hyperthyroid group, although this number did not reach statistical significance. The deficient parameters improved following treatment.
Little is known and there are scant data about the effects of hypothyroidism on human spermatogenesis and fertility. Investigators Dubin and Amelar in the early 1970s cited hypothyroidism as one of the syndromes of hormonal deficiency that rarely cause male infertility. Serum free testosterone and SHBG levels are normal or low . Also, in hypothyroid males the LH response to gonadotropin-releasing hormone seems to be blunted . Subclinical hypothyroidism does not seem to impact semen density, motility, or morphology .
Hypogonadism
By traditional epidemiologic methods, an estimated 1 in 200 men have abnormally low levels of testosterone . Hypogonadism is defined as a low total testosterone level (Food and Drug Administration [FDA] normal range, 300–1000 ng/dL) that can be associated with clinical findings such as decreased libido, infertility, anemia, mood changes, alterations in body hair distribution, and decreases in lean muscle mass and bone mineral density. Although men initially have 700 million Leydig cells, they lose approximately 6 million per year after age 20 years . Nieschlag and colleagues , however, did not find any significant differences in sperm counts or fertilizing capacity between younger men and men beyond their seventh decade. Hypogonadism can be separated further into primary and secondary forms. Primary hypogonadism is defined as a low serum testosterone level with normal or elevated LH. Serum FSH levels may be normal or elevated, as seen in patients who have Klinefelter’s syndrome, anorchia, or other iatrogenic causes.
Hypothalamic-pituitary axis abnormalities
Secondary hypogonadism can be categorized further into congenital forms such as idiopathic hypogonadotropic hypogonadism (IHH) ( Box 2 ) or acquired forms. IHH is defined as a low serum testosterone level associated with a low LH level with or without a low FSH level. An important caveat is that low LH levels do not always correlate with low FSH levels, and visa versa. Acquired forms of inappropriately low LH levels include hypopituitarism, hyperprolactinemia, estradiol excess, and noncongenital idiopathic causes. The clinical presentation and the diagnostic implications are based in part on whether this form of androgen deficiency is pre- or postpubertal.
Congenital idiopathic hypogonadism
- •
Anosmic (Kallman syndrome)
- •
Nonanosmic: normosmic idiopathic hypogonadotropic hypogonadism
- •
Fertile eunuch syndrome
Adrenal hypoplasia congenital
Genetic defects of the gonadotropin subunits
- •
Follicle-stimulating hormone-beta mutations
- •
Luteinizing hormone-beta mutations
- •
Mutations in leptin and leptin receptor genes
- •
Hypogonadotropic hypogonadism associated with other pituitary hormone deficiencies
- •
PROP-1 mutations
- •
HERS1 mutations
- •
Complex syndromes that include hypogonadotropic hypogonadism
- •
Prader-Willi syndrome
- •
Congenital spherocytosis
- •
Moebius syndrome
- •
Cerebellar ataxia
- •
Retinitis pigmentosa
- •
Kallmann syndrome is the anosmic form of IHH. Associated clinical features may include neurologic abnormalities (synkinesia, oculomotor deficits, deafness, mental retardation, and cerebellar defects,) unilateral renal agenesis, midline facial defects, and pes cavus. Kallmann syndrome is a common X-linked recessive form of human hypogonadotropic hypogonadism . In a recent report from Bhagavath and colleagues , KAL1 mutations were an uncommon cause of Kallmann syndrome in male patients, occurring in about 3.7% of all IHH males and in 6.3% of anosmic/hyposmic IHH males in their study population. IHH thus is probably far more common that previously expected, with patients who have Kallmann syndrome representing merely the “tip of the iceberg.”
Pituitary
Hypopituitarism is the partial or complete insufficiency of anterior pituitary hormone secretion and may result from pituitary or hypothalamic disease. The reported incidence (12–42 new cases per million population per year) and prevalence (300–455 per million population) probably are underestimated. Clinical manifestations depend on the extent of hormone deficiency and may be nonspecific (eg, fatigue, hypotension, cold intolerance) or more specific (eg, growth retardation secondary to insufficiency of growth hormone or impotence and infertility as a result of gonadotropin deficiency). A number of inflammatory, granulomatous, or neoplastic diseases as well as traumatic or radiation injuries involving the hypothalamic-pituitary axis can lead to hypopituitarism .
Recent observations
At the University of Illinois in Chicago, the authors recorded the incidence of hypogonadism in various diagnostic categories of patients presenting to an infertility clinic by retrospectively reviewing the charts of 120 consecutive patients. Inclusion criteria were diagnoses of NOA, obstructive azoospermia (OA), oligospermia defined by World Health Organization (fourth edition) criteria, and men who had normal semen analyses. Serum testosterone was measured by electrochemiluminescent immunoassay or turbulent flow liquid chromatography tandem mass spectrometry; FDA criteria defined hypogonadism as a morning serum testosterone level less than 300 ng/dL. Interestingly, the incidence of hypogonadism in men who had NOA was 45.0%. The incidence of hypogonadism was 42.9%, 35.3%, and 16.7%, respectively in men who had oligospermia, normal semen analyses, and OA ( Fig. 1 ). This subset of men who have OA may be considered a negative control because of the relatively exclusive nature of this anatomic condition. The fact that the incidence of hypogonadism in the OA group parallels that of the general population serves to validate the expectation that the OA group serves as a control. The lack of compensatory elevation of LH above 12.0 IU/L in the patient population suggested a form of hypogonadotropic hypogonadism. The finding that one third to nearly one half of the men who had NOA had an incidence of hypogonadism by FDA criteria supports the thesis that hypogonadotropic hypogonadism may be considerably more prevalent in the infertile male population than previously understood.






