and Andrea Bischoff1
(1)
Pediatric Surgery, Colorectal Center for Children Cincinnati Children’s Hospital, Cincinnati, OH, USA
24.1 Introduction
24.2 Historical Review
24.4 Pathogenesis
24.5 Genetics
24.7 Histologic Diagnosis
24.9 Early Management
24.10 Surgical Treatment
Electronic supplementary material
Supplementary material is available in the online version of this chapter at 10.1007/978-3-319-14989-9_24.
24.1 Introduction
Hirschsprung’s disease is a relatively common condition, well known to all pediatric surgeons. It is one of the most common causes of intestinal obstruction in the newborn. From the time of the original description by Harold Hirschsprung [1] and the proposal of the first rational surgical treatment by Orvar Swenson [2–4], multiple reports have been written in the international literature. The basic principles of the surgical treatment established by Dr. Swenson are still timely today. Yet, different modalities of treatment have been described aimed at trying to avoid morbidity related to the operation as well as trying to be as least invasive as possible. Simultaneously, there are a significant number of scientists looking into the genetics and pathogenesis of this condition, as well as the pathophysiology of the most feared sequelae and complication which is enterocolitis.
The reader will find this a rather unusual chapter. Since we are clinically active pediatric surgeons dedicated to the field of colorectal problems in children, we are privileged to receive a very high number of cases. We are also very impressed by the high number of patients referred to us, who were operated at other institutions and suffer from preventable complications consecutive to a poor surgical technique or lack of good clinical judgment. Interestingly, perhaps due to the nature of our center, we have treated, so far, more secondary cases than primary ones. Our database has information related with 328 cases; 102 cases were operated on by us primarily, 127 secondarily, and 99 were only medically managed. This has given us a unique insight into the problems affecting children who were born with Hirschsprung’s disease, problems which sometimes are not mentioned in the literature. Medical publications (articles and books) are frequently triumphant reports of good results and successful techniques. Generally speaking, the medical and surgical community has a tendency to avoid publications of failures, complications, and bad sequelae. In addition, very few pediatric surgeons follow their patients until adult life and, therefore, miss important feedback, which is related to the long-term sequelae and quality of life of those patients who we operated on in the pediatric age.
Since we personally do not have anything to contribute in terms of genetics and laboratory research related to this condition, we decided rather to share with the pediatric surgical community our clinical experience dealing with sequelae and complications, particularly those that we consider preventable, and the way to treat those problems. Therefore, the first part of this chapter will be a formal but rather short presentation of the main characteristics of the disease in the traditional academic fashion, which means a brief discussion about incidence, pathophysiology, clinical manifestations, radiologic studies, histologic findings, surgical treatment, and medical management of the sequelae. After that, we will expand on the preventable complications and the way to deal with those.
24.2 Historical Review
Harold Hirschsprung in 1886, at the Pediatric Congress in Berlin [1], described an infant who died with an enormously dilated colon. However, the cause of this was unknown. In 1691, Frederik Ruysch [5] also reported a case of a child who died that may have had the same condition. It was not until 1901 when Tittle for the first time mentioned the possibility that this condition could be provoked by an absence of ganglion cells of the distal rectosigmoid [6].
Frederick Treves in London in 1898 [7] actually performed the complete resection of a descending colon, sigmoid rectum, and anus on a 6-year-old girl. The patient survived, and Dr. Treves expressed his desire that she will recover her bowel control. That case represents the first case of cured Hirschsprung’s disease, although the surgeon did not know the intrinsic abnormality of the distal resected bowel. In 1900, Fenwick suggested that a possible cause of the disease was a spastic contraction of the lower end [8]. In 1907, Hawkins suggests that the condition could have a neuropathic origin [9].
Although not frequently mentioned, it was actually Dr. Alberto Dalla Valle from Parma, Italy, in 1920, who found that the distal narrow portion of the colon had no ganglion cells [10]. This remarkable finding was not mentioned for unknown reasons. For some years, a sympathectomy and daily enemas were the only treatment offered to these patients, with less than optimal results [11, 12].
In 1934, Arthur Hurst firmly believed that anal achalasia was the primary cause of this condition, as well as an imbalance of the parasympathetic and sympathetic innervations of the bowel. He also mentioned that the name “Hirschsprung’s disease should be discarded, as von Ammon gave a good description of two cases in 1842, 44 years before Hirschsprung’s of Copenhagen” [13].
The abnormal or absent peristalsis of the distal aganglionic bowel was described by Robertson and Kernohan in 1938 [14] and Tiffin et al. in 1940 [15], and subsequently, Zuelzer and Wilson in 1948 [16] correlated the functional disturbance of the rectosigmoid with the lack of ganglion cells. These findings were confirmed by Whitehouse and Kernohan in a study of 11 cases [17]. In 1948, Swenson in an experimental study [2] proposed a resection of the rectosigmoid with preservation of the sphincter for patients who suffer from this condition. In 1949, Swenson and Neuhauser (pediatric radiologist) published a very important paper on the etiology, diagnosis, and treatment of “congenital megacolon” [3]. After that, until the time of his retirement, Dr. Swenson accumulated an enormous experience in the management of this condition [4]. He also trained many surgeons who continued performing his operation until the present time.
Many surgeons around the world tried to imitate Dr. Swenson and reproduce his results in the treatment of this condition and found that sometimes the patients suffered from complications due to damage of pelvic structures or nerves. Because of that, in an attempt to reduce this morbidity, other surgical modalities were designed, but all of them were based on the same principle of resecting the aganglionic segment and pulling the normoganglionic bowel down. Most notable was the technique described by Duhamel in 1956 in France [18]; his technique will be described later in this chapter.
In 1959, Rehbein, in Germany published his experience in the treatment of Hirschsprung’s disease with an operation very similar to an anterior resection [19]. In other words, he left in situ a significant piece of aganglionic rectum. For that reason, the technique did not have many followers; yet surprisingly, there are many surgeons practicing it and claiming reasonably good results [20].
In Europe, Romualdi [21] in 1960, Ehrenpreis in 1961 [22], and Pellerin 1962 [23] published their experience with the resection of the rectosigmoid.
In 1964, Dr. Franco Soave from Italy [24] and Dr. Scott Boley from the United States [25], independently, submitted a paper to the pediatric section of Surgery (editor Dr. M. Ravitch). The technique that they both presented consisted in dissecting the rectosigmoid, in a surgical plane between the mucosa and the muscularis (endorectal), in order to protect the neighbor pelvic structures. The difference between both techniques was that Dr. Soave pulled the normoganglionic bowel down and left it exteriorized through the anus, for several days, after which he performed the anastomosis to the anus. Boley, on the other hand, was doing a primary anastomosis. For reasons unknown to us, the paper of Soave was published before the one of Boley. It is important to recognize that Boley’s paper was not presenting a modification to the Soave technique, but rather a modification to the Swenson technique [26].
More recently, the concept of minimally invasive surgery (laparoscopy) was applied. In 1994, Smith et al. [27] reported on one case who underwent a “laparoscopic Duhamel pull-through procedure.” Subsequently, Georgeson, in 1995, published his experience with 12 cases who underwent a “primary laparoscopic pull-through,” applying the endorectal dissection of the rectosigmoid performed through the abdomen and transanally [28]. This kind of technique was widely accepted.
In 1996, Saltzman et al. introduced a modification to the Soave technique, consisting in performing the entire mucosal dissection through a transanal approach as the abdominal incision was made. Their experience included 25 cases [29].
Dr. Luis de la Torre published his experimental work in rabbits, subjected to transanal, endorectal rectosigmoid resection [30]. His clinical experience with five cases, with Hirschsprung’s disease operated transanally, without laparotomy or laparoscopy, was published in 1998 [31]. Shortly after that, in 1999, Dr. Jacob Langer et al. published his experience with the transanal, one-stage Soave procedure [32].
When this operation is performed endorectally, basically it is the same principle of Soave, but done transanally and many times without opening or invading the abdomen. This revolutionary concept gained enormous popularity, and it is perhaps the most common surgical technique used at the present time. Yet, the basic principles proposed by Swenson must still be observed during this kind of operation. The fact that 85 % of the patients with Hirschsprung’s disease have an aganglionic segment that extends to the rectosigmoid, the need to open the abdomen or to use laparoscopy is reduced to about 20 % of the cases when using a transanal approach. The transanal approach is painless and when it is done correctly, has very good results [33].
24.3 Incidence, Inheritance, and Associated Anomalies
The estimated incidence rate was found to be 1 in 4,417 live births in what we thought was the most representative study of the subject [34]. It occurs four times more frequently in males in the typical form. In long-segment cases, this ratio is 1.5:1 (male/female). Familiar cases are three times more frequent in females and occur in approximately 4 % of the cases [35].
Associated defects occur in approximately 29 % of cases [34], mainly cardiovascular and gastrointestinal. Five percent of the cases may have Down syndrome [36]. Approximately 5 % of the cases may also suffer from a developmental disorder (Mowat-Wilson syndrome) [37]. Other associated problems include malrotation, ileal and colonic atresia, Dandy-Walker syndrome [38], and Ondine’s curse [39]. Finally a great interest developed recently about the “multiple endocrine neoplasia” (MEN) syndrome and its association with Hirschsprung’s disease [40]. This is mainly due to the ability to predict the risk for development of malignant tumors by genetic RET proto-oncogene analysis. This resulted in the recommendation to follow up these patients and the possibility to do a prophylactic thyroidectomy [40].
Many believe that Hirschsprung’s disease occurs more frequently in anorectal malformations as compared with the general population [41, 42], but we disagree with the concept. We believe that Hirschsprung’s disease is frequently erroneously diagnosed in patients who were born with anorectal malformation due to the fact that the most common sequela seen in anorectal malformation is constipation. Surgeons are educated and trained to suspect Hirschsprung’s disease in patients with constipation. It is not unusual, therefore, for them to overdiagnose this condition, and unfortunately, sometimes they treat it as such. We believe that Hirschsprung’s disease in anorectal malformations most likely occurs with the same frequency as in the general population.
24.4 Pathogenesis
The etiology and potential explanation of the possible origin and mechanisms of this fascinating condition has been the subject of an enormous number of studies and publications. It is beyond our area of expertise in the scope of this, mainly a practically oriented book, to analyze and discuss the multiple papers related with this subject. It is worth mentioning, however, the monumental work performed by Dr. Prem Puri [43–52].
Every year we read more new material coming from the laboratory of prominent research-oriented pediatric surgeons. The deeper they get in the investigation of the origin and the intrinsic mechanisms of this condition, the more complex it seems to be [53–61].
In 1967, Okamoto and Ueda [62] published an elegant paper, a study done with human embryos that allowed him to present his theory of the cranial-caudal migration of the neuroblast in the gastrointestinal tract. As time goes by and more research is performed, things seem to be more complex than previously thought.
From a more practical point of view, we can say that congenital megacolon (Hirschsprung’s disease) is characterized by a functional colonic obstruction usually manifested mostly during the newborn period. These patients do not have a real mechanical obstruction but rather have abnormal or absent peristalsis of the colon (usually the most distal portion). The part of the colon that has no ganglion cells does not have the necessary peristalsis to expel the stool, but rather is a spastic portion of the bowel. The aganglionic segment of the bowel is always located distally, and the length of the aganglionic segment varies from patient to patient. Typically, the aganglionic segment affects the rectosigmoid (Fig. 24.1). This type is known as “typical Hirschsprung’s disease” and represents about 67–82 % of the total number of cases. In about 15–25 % of the cases, the aganglionosis affects longer segments up to the splenic flexure or even the transverse or right colon (Fig. 24.1); this is called “long-segment Hirschsprung’s disease.” About 3–8 % of the patients suffer from aganglionosis of the entire colon including segments of the terminal ileum which is called “total colonic aganglionosis” [34]. Very rarely, patients are born with what is called “universal aganglionosis” which is mostly a lethal condition unless treated with intestinal transplantation. A condition known as “ultrashort-segment aganglionosis” (Fig. 24.1) is highly debatable; a strict rational, objective, scientific analysis of the available published material generates many doubts and skepticism about the existence of this condition. This will be discussed later.
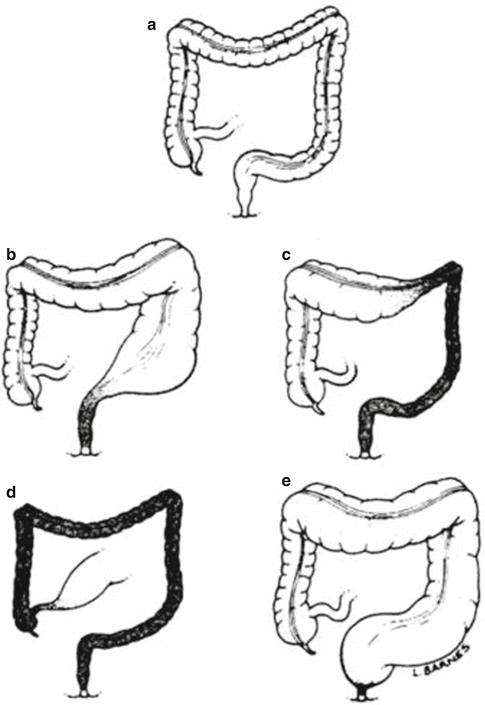
Fig. 24.1
Diagrams showing different types of Hirschsprung’s (a) Normal colon, (b) Classic type, (c) Long-segment, (d) Total colonic aganglionosis, (e) Ultra short type
The aganglionic segment in Hirschsprung’s disease has been described radiologically as the “narrow segment.” In reality, rather than narrow, it is a nondistended or spastic part of the colon. Proximal to the aganglionosis, the colon becomes extremely dilated (Fig. 24.1). At birth, the dilatation of the proximal colon may not be so severe (Fig. 24.2). However, as time passes, the dilatation gets worse, and the size discrepancy between the proximal dilated normoganglionic and the distal, non-dilated aganglionic segment becomes more conspicuous, eventually showing the typical radiologic image. The surgical literature frequently refers to the “transition zone” referring to that part of the bowel located between the dilated normal ganglionic and non-dilated aganglionic segment. The histologic study of the distal non-dilated bowel shows absence of superficial submucosa (Meissner), deep submucosal (Henle), and intramuscular (Auerbach) ganglion cells. In addition, there is a marked increase in size of prominent nerve fibers. The enzyme acetylcholine esterase activity is markedly increased. The ganglion cells act as a final common path for both sympathetic and parasympathetic activities. The aganglionic segment, therefore, suffers from spasm, lack of propulsive peristalsis, and contraction. In addition, characteristically these patients suffer from a lack of relaxation of the muscle that surrounds the lower part of the rectum described as an internal sphincter. This is a manometric finding. Recently, it has been proposed that nitric oxide, a neurotransmitter responsible for the inhibitory action elicited by the intrinsic enteric nerves, has a role in this condition. A lack of nitric oxide synthase (the enzyme required for nitric oxide production) may play a role in the pathophysiology [56–58].
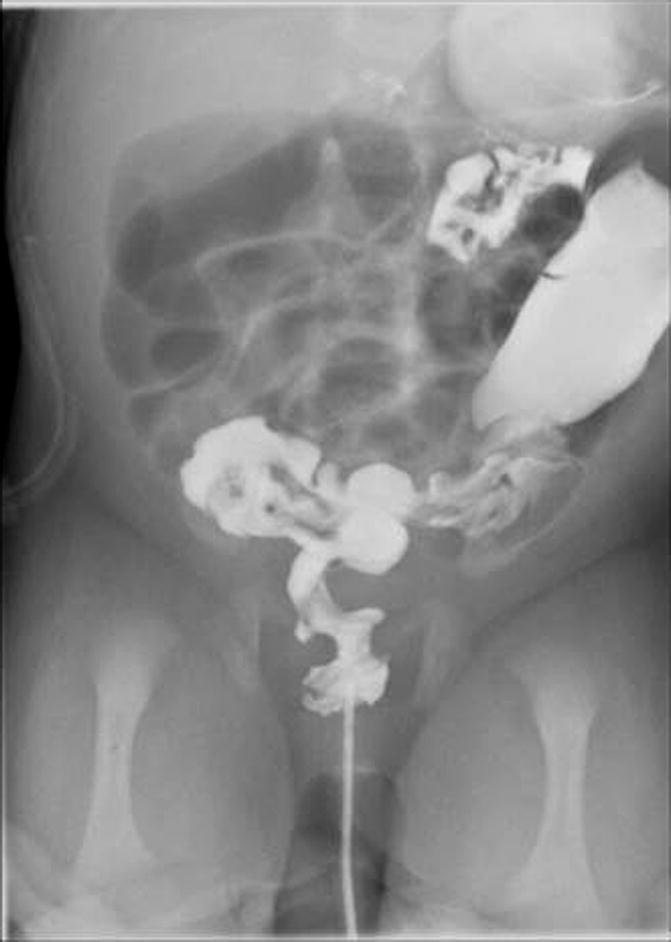
Fig. 24.2
Contrast enema showing the transition zone in a newborn. Contrast enema
Patients with Hirschsprung’s disease not only have manifestations of colonic obstruction but what makes this clinical picture different from other etiologies of colonic obstruction is the fact that proximal to the aganglionic segment, these patients frequently have a tendency to overgrow abnormal bacteria, specifically C. difficile, and they suffer from severe endotoxemia, a condition well known by pediatric surgeons as “Hirschsprung’s enterocolitis.” In other words, these babies not only have the pathophysiology and possible complications of lower intestinal obstruction, but in addition, they may become extremely sick and even die from the characteristic endotoxemia produced by enterocolitis.
24.5 Genetics
24.6 Clinical Manifestations and Differential Diagnosis
The first clinical manifestations of babies born with Hirschsprung’s disease are abdominal distention, vomiting, and the lack of passage of meconium in the first 24–48 h of life. Most neonatal units recognize the lack of passage of meconium in the first 24–48 h as a very suggestive sign of Hirschsprung’s disease. Every baby who has not normally passed meconium should be thoroughly investigated for the possibility of having Hirschsprung’s disease. There is a small group of patients born with Hirschsprung’s disease that do not have very obvious clinical manifestations at birth; they grow up and are diagnosed later in life. This group of patients most likely belongs to a category of Hirschsprung’s disease that we consider benign. In our experience, they rarely have episodes of enterocolitis, and when they are operated on, the results are better than those obtained in babies who have very early manifestations [72].
Characteristically, newborn babies with Hirschsprung’s disease look lethargic and the abdomen is distended. However, rectal stimulation either with a digital exam or with a catheter frequently results in an explosive and massively deflating passage of liquid stool and gas, which dramatically improves the baby’s condition. The baby may then show signs of being hungry and even eat, only to have the same symptoms return hours later and to repeat the cycle when the baby has a rectal examination. The liquid stool in these babies is characteristically fetid. Early detection of this dangerous stage of the disease is extremely important. These babies need aggressive resuscitation with intravenous fluids, fasting to avoid exacerbation of the symptoms, and a prompt and efficient way to decompress the colon. The most expedient and easy way to decompress the colon is using a rather large (no. 18 or no. 20 French) tube passed through the rectum and irrigate with saline solution. The liquid stool then comes out through the lumen of the tube, decompressing the baby’s abdomen. This is repeated several times during the day and allows the baby to improve his symptomatology significantly. The administration of intravenous metronidazole is also recommended to fight the bacterial overgrowth. When the colon is not adequately decompressed, these babies are at risk of dying from enterocolitis, endotoxemia, and hypokalemia or they may suffer perforation of the colon. The most common site of perforation is the cecum or the appendix. In addition, they may have pneumatosis intestinalis or develop pericolic abscesses. When this condition goes unrecognized, the mortality ranges between 25 and 30 % [73]. Some authors even report 50 % mortality for untreated patients during the first year of life [74].
The diagnosis of Hirschsprung’s disease is strongly suggested by the clinical symptomatology. A contrast enema reinforces the clinical suspicion, and finally, the diagnosis is confirmed by a rectal biopsy showing absent ganglion cells, the presence of hypertrophic nerves, and increased activity of acetyl cholinesterase. An abdominal plain x-ray film shows enormously dilated loops of bowel (Fig. 24.3). Most pediatric radiologists and experienced clinicians agree that it is extremely difficult to differentiate in a plain abdominal film in a newborn baby a distended colon from a distended small bowel. That may be possible in adults but is almost impossible in newborn babies. However, very few neonatal conditions give an image of such an enormous dilatation of loops of bowel. The next step after we see the plain abdominal x-ray film is to perform a contrast enema. Characteristically, the study shows a non-dilated portion, usually rectosigmoid, followed by a transition zone, and more proximately a dilated segment of the colon. These characteristic changes (Fig. 24.2) may not be present or may not be very obvious in the newborn period. As time goes by, the functional obstruction produces more proximal dilatation, and months or years later, the characteristic radiologic picture of a giant proximal megacolon with a narrow distal segment becomes very clear (Fig. 24.4). It is important to remember that in dealing with intestinal obstruction of the newborn, the only condition that has symptoms of intestinal obstruction and a dilated colon is Hirschsprung’s disease. All the others have characteristically a nonused colon (microcolon). An exception could be observed in babies with necrotizing enterocolitis, but the other symptoms and signs make this easy to differentiate from Hirschsprung’s disease.
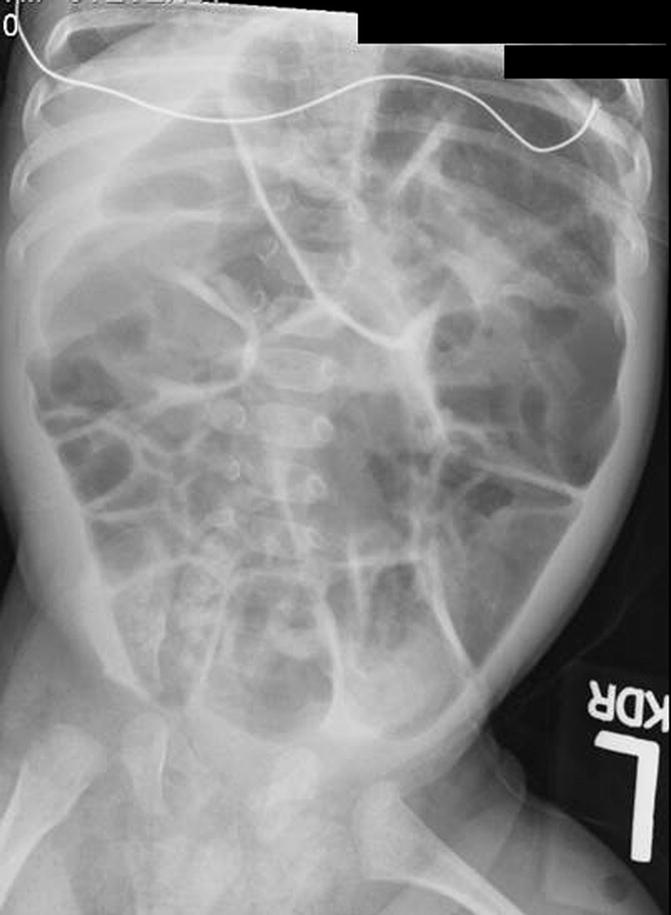
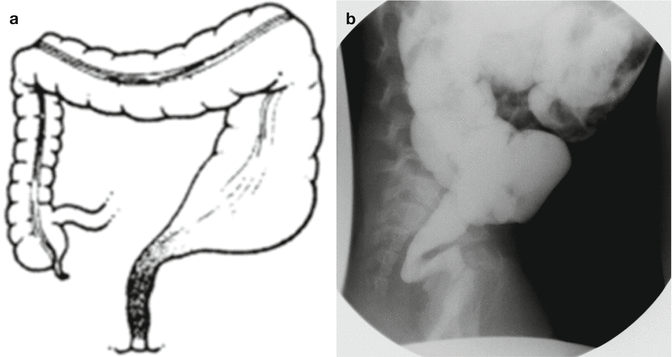
Fig. 24.3
Abdominal x-ray film of a newborn baby with Hirschsprung’s disease
Fig. 24.4
Characteristic colonic changes in Hirschsprung’s. (a) Diagram. (b) Contrast enema
Some newborns have a very obvious, radiologic image characteristic of Hirschsprung’s disease. In others, however, the transition zone and the proximal dilated portion are not so clear, yet together with the clinical manifestations allow us to make a diagnosis of Hirschsprung’s disease (Fig. 24.2). When the contrast study has been done, another characteristic of these babies is their incapacity to expel the contrast material 24 h after the study. This makes the diagnosis of Hirschsprung’s more likely.
We, like others [75], use and recommend a contrast enema as an initial diagnostic tool. We believe that with the combination of a good index of suspicion, clinical experience, contrast enema, and rectal biopsy read by an experienced pathologist, we can offer a safe management to these patients. We do not remember, from our large practice a false-positive or false-negative case.
We recognize that many times, the symptoms can be rather equivocal; in addition, sometimes the images obtained with the contrast enema may not be characteristic. Also, we had cases in which the suction rectal biopsies did not obtain good specimens. In such cases, we take a full rectal biopsy, which finalizes the diagnostic work. We are aware of the fact that many surgeons value the rectal manometry for the diagnosis of Hirschsprung’s [76–82]. We have no experience with that diagnostic modality.
24.7 Histologic Diagnosis
Very significant advances occur in the knowledge of the histologic abnormalities present in this disease [83–87]. Sometimes, we surgeons feel overwhelmed by the variety of terms and abundance of new findings. We keep trying to familiarize with a variety of terms like VIP (vasoactive intestinal peptide), adenylate cyclase-activating polypeptide (PACAP), gastrin-releasing peptide (GRP), calcitonin gene-related peptide (CGRP), substance P (SP), enkephalins and galanin-immunoreactive nerve fibers, neuropeptide Y (NPY)-containing nerve fibers, and calretinin.
Despite all the advances, the fundamental basis for the diagnosis of Hirschsprung’s disease still depends on a skillful pathologic analysis and the use of hematoxylin and eosin stains, to identify the presence or absence of ganglion cells and hypertrophic nerve trunks. Acetylcholinesterase (AchE) histochemistry on frozen sections and immune histochemistry for ganglion cells are useful ancillary techniques, but they do not replace the H&E methodology. Adequate sampling, extensive sectioning, and an experienced pathologist are the most important aspects of a good histologic diagnosis [88].
Different clinicians give a different value to the diagnostic tools for Hirschsprung’s disease. Some believe very much in the rectal mucosal suction biopsy, and that is all they do to make the diagnosis. Others prefer a full-thickness rectal biopsy. In certain institutions, there are experienced pathologists dedicated to the diagnosis of Hirschsprung’s disease by a suction rectal mucosal biopsy. At other institutions, pathologists do not have experience with this; they insist that the tissue obtained with suction rectal biopsy is not enough for a definitive diagnosis, and they demand a full-thickness specimen to make a reliable diagnosis. We try not to be dogmatic in this chapter concerning which diagnostic tool is more valuable than others. We rather believe that it depends on the specific surrounding circumstances where the baby is born and the experience of the surgeon, the neonatologist, the pathologist, and the radiologist. As a matter of fact, in one of the institutions where we worked in the past, there was only one pathologist that had enough experience in Hirschsprung’s disease to make a reliable diagnosis. Therefore, we only operated on Hirschsprung’s disease patients when that particular pathologist was present. The main message that we would like to give to the pediatric surgical community is that we should not assume that every pathologist can make the diagnosis of Hirschsprung’s disease. For instance, the diagnosis based on frozen section is reliable for certain specific experienced pathologists but not for others. Therefore, the surgeon must be ready to follow a different surgical strategy in the management of these patients depending on his or her surrounding circumstances. In some countries that we have visited, there are no pathologists available sometimes for several days, and therefore, that requires a different surgical strategy. In some places, the surgeons tend to believe only in the increase of the activity of acetylcholine esterase rather than on the absence of ganglion cells. Most pathologists all over the world agree, like us, that the basis for the diagnosis should be the absence of ganglion cells and the presence of hypertrophic nerves.
24.8 Differential Diagnosis
The most common condition that must be differentiated from Hirschsprung’s disease is the meconium plug syndrome. The expulsion of the meconium plug with disappearance of the symptoms allows Hirschsprung’s to essentially be ruled out. Meconium ileus is an early manifestation of cystic fibrosis, and therefore, one expects to find the other stigmata of cystic fibrosis including intestinal obstruction, an image of “ground glass” representing inspissated meconium in the small bowel, respiratory symptoms, lack of air-fluid levels, and response to the specific treatment to try to liquefy the inspissated meconium.
One very unusual condition, called small left colon syndrome, occurs when the narrow portion of the colon on the contrast enema usually reaches the splenic flexure. Symptoms usually disappear after the contrast study has been done and resolve spontaneously after several weeks. The mothers of these babies frequently have maternal diabetes.
Several other conditions are associated to hypomotility of the colon and may mimic Hirschsprung’s including hypothyroidism and effects from opiate or magnesium sulfate, transmitted from the mother.
24.9 Early Management
We recommend the administration of metronidazole as soon as we suspect the diagnosis of Hirschsprung’s disease; however, we believe that the most valuable maneuver that actually saves many lives in babies with Hirschsprung’s disease is colonic irrigation with saline solution (Animation 24.1).
Unfortunately, in many institutions, the term “colonic irrigation” is confused with an enema. An enema consists of the introduction of a specific volume of a fluid into the colon in order to provoke a bowel movement. By definition, patients with Hirschsprung’s disease have poor peristalsis, incapacity to expel fluids, and therefore, enemas are contraindicated as they may be retained and aggravate the symptomatology. On the other hand, irrigation means the passing of a tube through the rectum, large enough to allow the expulsion of liquid stool through the lumen of the tube itself. The term “irrigation” comes from the idea of passing, through the lumen of the tube, small volumes of saline solution, 10 mL at a time, in order to clear the lumen of the tube to allow decompression of the colon. The tube is well lubricated and is passed through the rectum, which allows an explosive decompression of most of the colon. After this first episode, the nurse or the doctor passes small volumes of saline solution through the tube and keeps moving the tube back and forth and rotating it, trying to get into the pockets of gas and liquid stool (See Animation 24.1 showing a “Colonic Irrigation”). The benefit of this irrigation is very obvious from the moment it is done. In fact, it may save the baby’s life. Even when this explanation of the difference between an enema and irrigation sounds obvious, we have been impressed by the frequency with which these two terms are confused even in well-reputed institutions and how often irrigations are neglected in the initial management altogether.
24.10 Surgical Treatment
The main goal in the treatment of patients with Hirschsprung’s disease is to resect the aganglionic segment and pull down normoganglionic bowel that must be anastomosed to the rectum above the anal canal, preserving the part of the bowel where the sensitivity resides and preserving the sphincter mechanism (anal canal). All techniques, regardless of its specific maneuvers, must observe these basic principles.
There is some controversy about how to treat these babies. Should we perform this operation primarily? Or should we open a colostomy first?
Rather than recommending a specific type of approach to this condition, we believe that the surgical approach depends on the specific circumstances that surround the patient as well as the surgeon. A primary operation for Hirschsprung’s disease done in the newborn requires experience, a sophisticated surgical environment, good anesthesia, good intensive care, good pathology, a sterile environment, as well as possibilities to provide parenteral nutrition to the baby. This, we take for granted in the United States; yet, it is not available in many other countries, and therefore, different strategies must be followed to allow these babies to survive.
Traditionally, these patients received a colostomy at birth, usually a right transverse colostomy followed later in life by an abdominoperineal resection of the aganglionic segment with pull-through of the normoganglionic bowel and a third operation consisting of a colostomy closure. Subsequently, another way to approach these patients consisted in opening what is called a “leveling colostomy,” meaning to open the colostomy in the transition zone; later on, in a second operation, the aganglionic segment was resected and the normoganglionic colostomy site was pulled down to be connected to the rectum. This was known as a “two-stage approach.” Finally in 1980, Dr. Henry So, in Long Island, New York, published his experience with an endorectal pull-through without colostomy in neonates with Hirschsprung’s [89]; soon after, that modality of treatment was adopted by many surgeons [90–101]. Other modalities include a laparoscopic primary approach and, more recently, the primary transanal approach [30].
We believe that there are many ways to solve a problem, and all of them can be equally good. Regardless of the therapeutic approach used, the fundamental goals of treatment should be to resect the aganglionic segment and to pull down a normoganglionic piece of colon without causing any harm to the pelvic organs, or their nerves, and preserving intact the anal canal. This can be done in a single, primary operation or with two or even with three operations, depending on the specific circumstances of the patient, the surgeon’s experience, and the surrounding environment. The main goal can also be achieved using different techniques, laparoscopically or open. It is fashionable among surgeons to talk about the “gold standard” when referring to the best way to treat a specific condition. It is also very common for pediatric surgeons to compete trying to perform these operations earlier and earlier in life and using the least invasive technique, with a minimal length of stay in the hospital and minimal cost. Looking at our experience in dealing with patients who came to our institution after a failed attempted repair, or after suffering catastrophic preventable complications, lead us to believe that it is not so important which treatment modality was used. What is really important for the patient is to be operated by a meticulous, experienced surgeon, using the treatment modality with which he or she feels most comfortable and with which the aforementioned goals can be achieved.
Clearly, it is ideal to repair a defect as early as possible, in the least invasive way, and in a single operation, but far more important than that, to avoid catastrophic, preventable complications such as neurogenic bladder, fecal incontinence, dehiscence, retraction, rectovaginal fistula, and rectourethral fistulas.
24.10.1 The Authors’ Approach
If a baby is born nearby our institution or is brought very early with symptoms consistent with Hirschsprung’s disease, we perform a contrast enema and a suction rectal biopsy. Once we establish the diagnosis, assuming it is a full-term baby with no concerning associated defects and is clinically stable, we insert a central line (PICC line) and keep the patient with nothing by mouth receiving parenteral nutrition. We have learned that these patients do not need a nasogastric tube provided one keeps the colon decompressed. The babies vomit when they have a distended colon and have enterocolitis, but the decompression of the colon stops the vomiting. A program of irrigations three times per day and intravenous metronidazole is started. The goal of the irrigations is to avoid enterocolitis and to maintain the colon decompressed and the abdomen flat. Gradually, over the period of a few days, we can see that what used to be fetid liquid stool becomes less and less so more clear and eventually becomes bilious and odorless. Occasionally (rare in our institution), one can see a baby suffering from severe enterocolitis, endotoxemia, and shock coming to the hospital. Under those circumstances, we understand that may be justified to open a colostomy to save the baby’s life. At least in the environment where we work, this particular situation occurs very rarely. Newborn babies come to our hospital looking sick, yet, they respond well to the management described and can be operated primarily after a few days of medical management (irrigations and metronidazole).
At this point, with no evidence of enterocolitis, a nondistended colon, and the baby in good condition, we perform a primary transanal repair. Based on the fact that 85 % of the patients have an aganglionic segment that extends only to the rectosigmoid, we believe that it is necessary to open the abdomen or to perform a laparoscopy in approximately 15 % of the cases. Some surgeons advocate the use of laparoscopy in all cases, to take biopsies to determine the extent of the aganglionosis. We believe that it is not necessary to follow that approach. Transanally, as we progress in our dissection, we can take full-thickness biopsies every 5 cm until we reach the normoganglionic bowel. If the contrast study gives no clue as to the location of the transition zone or if we are suspicious of total colonic Hirschsprung’s, starting with laparoscopy and biopsies may be appropriate.
We perform a full-thickness (like Swenson) transanal (de la Torre) dissection. Based on a large experience in the treatment of anorectal malformations, we have learned to dissect the rectum, keeping our plane of dissection as close as possible to the bowel wall without injuring any nerves and/or pelvic organs. In other words, we do not feel the need to do this dissection using an endorectal, submucosal technique. As we gain length in the dissection, we keep taking biopsies every 5 cm, sending them to pathology. Once we have reached the normal ganglionic bowel, we still go 5 cm or more, proximally. If the normal ganglionic bowel is very dilated, we continue pulling down more colon. In other words, we try to resect not only the aganglionic segment but also as much of the dilated normal ganglionic colon as possible. After the resection, a two-layer anastomosis is done 2 cm above the pectinate line. If, in the process of dissecting the rectum, we find that it is becoming technically more demanding and difficult to continue the dissection because we already have passed the sigmoid and reaching the descending colon becomes extremely difficult, then we decide to go into the abdomen either laparoscopically or opening the abdomen to free the descending colon, the splenic flexure, or even the transverse colon.
The transanal approach is a very important significant contribution in the therapy of Hirschsprung’s disease. We believe that both Drs. de la Torre and Langer should be commended for this contribution. Yet, we have serious concerns that this technique may1 not be very reproducible. In other words, when this technique is not done correctly, serious damage may be provoked to the sphincter mechanism and to the anal canal. We have seen videos and photographs of transanal operations presented in different meetings, showing images with rather aggressive, less-than-optimal maneuvers that we believe may provoke fecal incontinence. Because of that, we want to emphasize what we consider the crucial steps in the surgical technique of the transanal resection of the rectosigmoid (see attached DVD).
We use a Lone Star2 retractor to perform this operation [102, 103]. This retractor provides an excellent exposure combined with minimal invasiveness. The baby is placed in the prone position with the pelvis elevated. We believe that there is no need to operate on these babies in lithotomy position, which is a reminiscent of adult surgical techniques. With the patient in prone position, the surgeon works more comfortably in a horizontal field without losing instruments and can perform the dissection easier, and this position is much easier on the assistant and the scrub nurse. The eight hooks of the Lone Star retractor are placed in a symmetric and radial way at the anal margin in order to be able to see the pectinate line (Fig. 24.5). Subsequently, all the hooks are replaced deeper, taking the rectal wall above the pectinate line (Fig. 24.6) so that it is hidden. At that point, we know that what is exposed is only rectal mucosa. We are now sure that the entire anal canal, plus at least 1 cm of rectal mucosa, has been circumferentially folded, retracted, and protected. What we see deep in our field (Fig. 24.6) is only rectal mucosa. This way we can be very accurate in measuring the distance between the pectinate line and the beginning line of our resection. Many authors talk about initiating the resection 5 or 10 mm above the pectinate line. We prefer to go 2 cm more proximal for the reasons that we will explain when we talk about complications from these procedures. Multiple 5-0 or 6-0 silk circumferential stitches are placed taking the rectal mucosa (Fig. 24.7). These stitches will provide uniform traction. We use a delicate needle-tip cautery and make a full-thickness circumferential incision of the rectal wall, peripheral to the multiple silk stitches (Fig. 24.8). We use the cautery in cutting mode when cutting and coagulation mode to fulgurate specific vessels. Peripheral to the full-thickness wall of the rectum, one finds a funnel-like skeletal muscle that contracts every time we touch it with the cautery. We should not violate that wall of skeletal muscle. If we go through that, we find characteristic ischiorectal fat that should alert us that we are too far away from the correct plane and therefore at risk of injuring other structures. The dissection of the anterior rectal wall in male patients must be performed very meticulously. It is perfectly valid to go submucosally in this particular area to guarantee that we are not injuring the prostate and the posterior urethra. Likewise in females, the posterior vaginal wall must be protected. If the dissection is performed correctly, one finds that dissecting the rectum in a full-thickness fashion allows us to work in a bloodless field only burning the extrinsic blood vessels of the rectum. This represents a much less bloody field than when using a submucosal dissection. When one is performing this dissection in the right plane, the dissection is performed most of the time outside the level of the retractor. In other words, we keep dissecting and pulling the rectum mostly outside the anus; we try not having to work in a deep field unless we are dealing with a reoperation with a lot of fibrosis in the pelvis. Every 5 cm of length that we gain, we take a full-thickness biopsy that is sent to pathology (Fig. 24.9). Obviously, this approach is safe when one is working in an environment with a pathologist who has experience with frozen sections. In about 80 % of the cases, we reach the normoganglionic bowel without the need to open the abdomen or doing laparoscopy. Yet, we go at least 5 cm above the site of the biopsy that showed ganglion cells; we also resect as much of the dilated normoganglionic bowel as possible through the same transanal approach. A two-layer anastomosis between the normoganglionic bowel and the rectum is performed, 2 cm above the anal canal (Fig. 24.10). The first layer of the anastomosis takes the external or serosal part of the colon and sutures it to the peripheral tissues above the edge of the resection of the rectal mucosa. This first layer of sutures is performed before the resection of the colon. Figure 24.11 shows the resection of the colon. In the second inner layer, more emphasis is placed in being sure that the mucosal edges come together. This suture is performed with 6-0 Vicryl (Fig. 24.12). During the dissection, we assist ourselves with the use of small pediatric malleable retractors and a long, narrow Deaver retractor. During this dissection, we work following the principle of triangular exposure. We pull on the multiple silk rectal stitches in one direction, use the suction tube to improve the exposure, and 1 or 2 narrow retractors are introduced in order to see exactly what we are doing. We are very careful to not excessively stretch the anal canal. We believe that serious damage may be inflicted when the surgeon stretches the anal canal excessively in order to have better exposure.
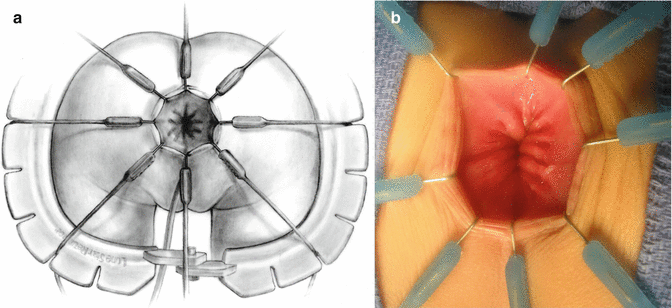
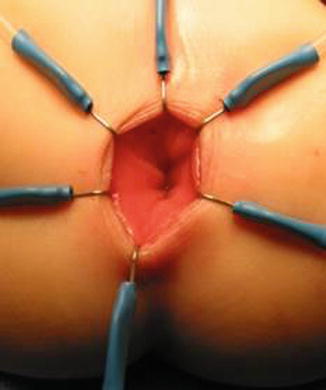
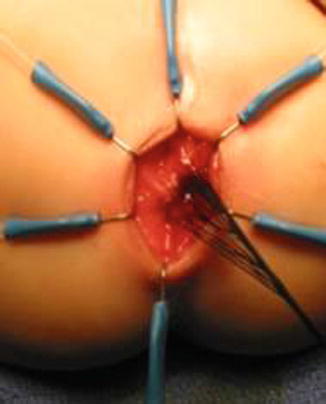
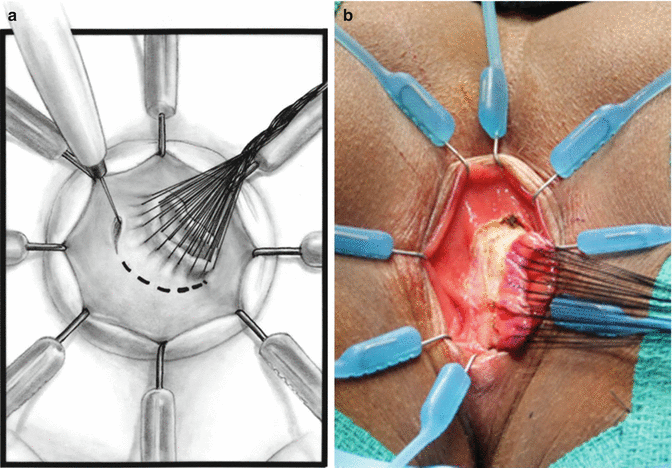
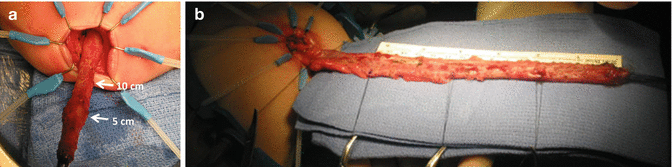
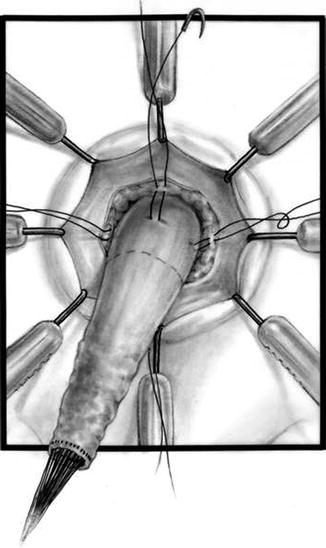
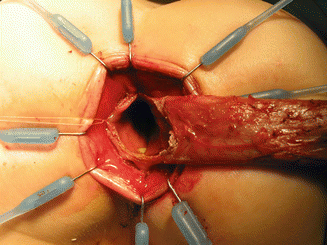
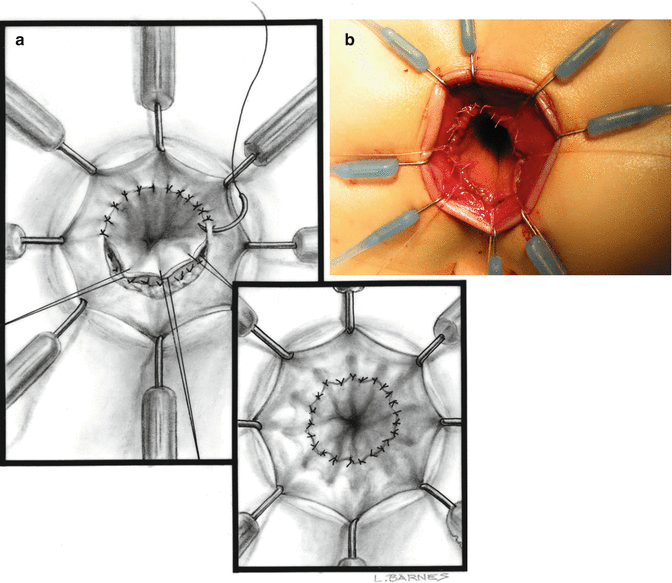
Fig. 24.5
Eight hooks in place. (a) Diagram. (b) Intraoperative picture
Fig. 24.6
Eight hooks placed deeper – only rectal mucosa is exposed. The anal canal is protected (folded) by the hooks of the retractor
Fig. 24.7
Multiple silk stitches
Fig. 24.8
Circumferential incision, peripheral to the silk stitches. (a) Diagram. (b) Intraoperative picture
Fig. 24.9
Biopsies taken every 5 cm (a) 15 cm dissected, (b) 20 cm dissected
Fig. 24.10
Diagram showing the deep layer of the anastomosis
Fig. 24.11
Resection of the colon
Fig. 24.12
Superficial layer of stitches (inner layer). (a) Diagram. (b) Operative picture
In about 20 % of patients, the dissection continues, but we cannot reach the normoganglionic bowel. The dissection becomes more difficult and risky because we are trying, through the anus, to cauterize mesenteric intraperitoneal vessels that may retract and bleed into the peritoneal cavity. Under these circumstances, we go into the abdomen, either laparoscopically or by laparotomy. This is easily done because we perform these operations using what we call “total body preparation.” We use this type of preparation modality in all of our abdominoperineal procedures. The reader can go to Chap. 7 to see the details of a total body preparation. When necessary, the baby is turned to supine position and laparotomy or the laparoscopic approach is performed. In order to avoid the leak of CO2 from the peritoneal cavity, packing gauze can be used in the anus. Laparoscopically, the descending colon is detached from its normal (left gutter) attachments as well as the splenic flexure. This maneuver is usually enough for the normoganglionic bowel to reach the anus and to perform a successful tension-free anastomosis. If there is not enough length, we have to detach the hepatic flexure to gain extra colonic length. Sometimes the right colon must be de-rotated for the hepatic flexure to reach the anus. Rarely, one is confronted with the case of a total colonic aganglionosis which should have been suspected preoperatively. This will be discussed later.
If the decision is made to go into the abdomen through a laparotomy, we prefer a midline incision. In dealing with colorectal problems, we favor the midline abdominal incisions in order to preserve both sides of the abdominal wall in the event that the patient requires a stoma. Transverse incisions invade the area of the stomas, and the patients may end up with a stoma located too close to an incision, which is a serious inconvenience, since it may interfere with the placement of the stoma appliance.
At the end of the procedure, most of the time, the patient stays without a protective colostomy, provided we feel comfortable about the blood supply of the pulled-through bowel as well as the tension of the anastomosis. We believe that strictures that occur at the anastomosis site are due to poor surgical technique and frequently ischemia of the bowel or excessive tension. If it happens that the patient requires a laparotomy or laparoscopy, the surgeon must be very careful in observing the blood supply of the colon in order to be sure that they ligate or cauterize the right vessels to avoid ischemia of the distal bowel. The anastomosis should not be done under tension. When there is any question about the viability of the bowel as well as the tension, a protective colostomy is indicated.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


