Fig. 37.1
Colorectal cancer viewed broadly
Non-syndromic hereditary colorectal cancer refers to familial clustering that does not fit criteria for the definition of a syndrome and no germ line mutation is found.
Syndromic hereditary colorectal cancer is more important, however, because of the extremely high level of risk associated with it and because it is relatively easier to identify.
Syndromic Hereditary Colorectal Cancer
A syndrome is a condition characterized by a constellation of symptoms, signs, and associations that go together so that the presence of one feature may alert the clinician to the presence of others.
Hereditary colorectal cancer syndromes can be broadly separated into those that are associated with multiple polyps (the hereditary polyposis syndromes) and those that are not (hereditary nonpolyposis colorectal cancer (HNPCC)). These syndromes and their definitions are listed in Table 37.1.
Table 37.1
Hereditary colorectal cancer syndromes
Polyposis syndromes
Phenotypic definition
Genotype
Familial adenomatous polyposis
Attenuated: <100 synchronous adenomas
Dominant inheritance of germ line mutation in APC
Mild: <1,000 synchronous adenomas
Severe/profuse: >1,000 synchronous adenomas
MYH-associated polyposis
Attenuated/mild polyposis
Recessive inheritance: biallelic mutations of hMUTYH
Hyperplastic polyposis
>20 hyperplastic polyps of any size or location
Unknown
>50 hyperplastic polyps proximal to sigmoid, 2 >10 mm
Any number of hyperplastic polyps with a family history of hyperplastic polyposis
Hamartomatous polyposes
Two of the following criteria:
1. Peutz–Jeghers syndrome
Mucocutaneous pigmentation
Dominant inheritance of germ line mutation in STK11
Gastrointestinal Peutz–Jeghers polyps
Family history of Peutz–Jeghers polyposis
2. Juvenile polyposis coli
>4 juvenile polyps in the colorectum
Dominant inheritance of germ line mutation in SMAD4 or BMPR1
Any number of juvenile polyps and a family history of juvenile polyposis
3. PTEN tumor hamartoma syndromes
Dominant inheritance of a germ line mutation in PTEN
(a) Cowden’s syndrome
International Cowden Consortium Criteria
(b) Bannayan–Riley–Ruvalcaba syndrome
(c) Proteus syndrome
Nonpolyposis colorectal cancer
Lynch syndrome
Dominant family history, microsatellite-unstable (high) colorectal cancer, young age of onset
Dominantly inherited germ line mutation of DNA mismatch repair gene: hMLH1, hMSH2, hPMS2, hMSH6
Familial Colorectal Cancer Type X
Dominant family history, microsatellite-stable tumor
Unknown
All of them confer an enhanced risk of colorectal and extracolonic cancers on affected patients and demand a sophisticated knowledge of genetics and medical and surgical treatment from caregivers.
The Polyposis Syndromes
The Adenomatous Polyposes
Familial Adenomatous Polyposis
Familial adenomatous polyposis (FAP) is an autosomal, dominantly inherited condition due to a germ line mutation of APC, which occurs with a frequency of about 1:10,000 live births.
About 22 % of germ line APC mutations occur “de novo,” meaning that there is no family history of the syndrome.
Inactivating mutations of this tumor suppressor gene result in a generalized disorder of growth regulation with a range of clinical manifestations, principally the formation of multiple gastrointestinal adenomas and carcinomas.
FAP is thought to account for between 0.05 and 1 % of all colorectal cancers.
Patients with a diagnosis of FAP and their family should be referred to a polyposis registry.
Polyposis Registries
The aim of polyposis registries is to provide counseling, support, and clinical services for families with FAP.
This includes thorough pedigree analysis and identification of at-risk family members, who are offered genetic testing and clinical surveillance.
Those shown to be affected can be offered prophylactic surgery.
Some registries also coordinate postoperative surveillance and provide a focal point for education, audit, and research.
Observational studies suggest that the introduction of registries, together with the use of prophylactic surgery, has led to increased life expectancy and a dramatic reduction in the incidence of colorectal cancer in FAP.
Features of FAP
The Large Bowel. The cardinal manifestation of FAP is the development of over 100 colorectal adenomatous polyps, one or more of which inevitably progress to carcinoma if not removed (Fig. 37.2).
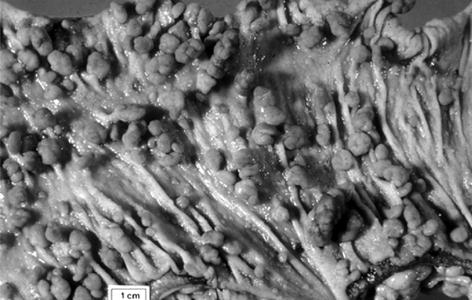
Fig. 37.2
The large bowel in classical familial adenomatous polyposis
Polyps usually appear in adolescence, with colorectal cancer diagnosed at an average age of about 40 years.
The severity of the colorectal polyposis is an important determinant of treatment and is used to define the pattern of FAP.
Patients with less than 100 adenomas are classified as having attenuated FAP, and this phenotype overlaps significantly that of MYH-associated polyposis (MAP).
Patients with 100–1,000 adenomas have classical FAP, while those with >1,000 adenomas have profuse FAP.
Polyposis severity is partly a reflection of the location of the APC mutation and partly due to unidentified modifying factors. The “hotspot” mutation at APC codon 1309 is reliably associated with profuse polyposis.
Genetics
The APC Gene. APC is a large gene on chromosome 5q21 (q = the long arm).
It is a key (gatekeeper) gene in colorectal carcinogenesis and is mutated in a majority of sporadic colorectal cancers.
Over 820 different germ line APC mutations causing FAP have been identified, almost all resulting in truncation of the APC protein. Mutations have been found between codons 168 (exon 4) and 2839 (exon 15), but most are between codons 168 and 1640 (exon 15) in the 5′ half of the coding region, with a particular concentration at two “hotspots,” codons 1061 and 1309.
The APC Protein. APC is expressed in all organs, but the mRNA is found at particularly high levels in normal colonic mucosa.
In many epithelia, APC is only found when cell replication has ceased and terminal differentiation is established.
The 300 kDa APC protein is found in the cytoplasm and has sites of interaction with a range of other proteins, including β-catenin and the cytoskeleton. It plays a central role in the highly conserved Wnt signaling pathway, which is involved in the normal development of three-dimensional structures and is abnormally activated in some malignancies.
APC binds and downregulates cytoplasmic β-catenin, preventing its translocation to the nucleus. Abnormal APC fails to do this so that β-catenin is free to enter the nucleus and form a complex, which results in specific transcription of cell cycle stimulating DNA sequences, and hence cell proliferation.
Genotype–Phenotype Correlation in FAP
There is evidence of correlation between the position of the germ line APC mutation (genotype) and some aspects of phenotype (Fig. 37.3).
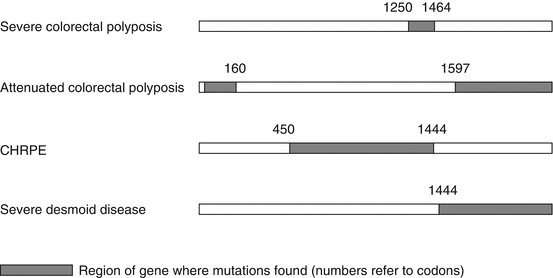
Fig. 37.3
Schematic representation of the APC gene showing genotype–phenotype correlations
Mutation at codon 1309 is associated with profuse polyposis and between codons 1250 and 1464 with earlier onset of, and death from, colorectal cancer.
Mutations located 5′ of codon 160 and 3′ of codon 1597 are associated with mild or attenuated colonic polyposis, accounting for about 10 % of those affected.
Some extracolonic manifestations have also been associated with mutations at certain sites, although not upper gastrointestinal polyposis.
Congenital hypertrophy of the retinal pigmented epithelium (CHRPE) occurs only with mutations between codons 450 (exon 9) and 1444.
The association of desmoid disease with germ line APC mutations 3′ of codon 1444 can be clinically important, although identical APC mutations may be associated with diverse phenotypes, suggesting that other genetic modifiers are involved.
Clinical Variations of FAP
Extracolonic Manifestations
The extracolonic manifestations of FAP are shown in Table 37.2.
Table 37.2
Extracolonic features of familial adenomatous polyposis
System
Feature
Frequency (%)
Upper gastrointestinal tract
Upper gastrointestinal adenom
95
Upper gastrointestinal carcinoma
5
Fundic gland polyps
40
Connective tissue
Osteomas (especially jaw)
80
Desmoid tumor
15
Dental
Unerupted and supernumerary teeth
17
Cutaneous
Epidermoid cysts
50
Endocrine
Adrenocortical adenomas
5
Papillary thyroid carcinoma
1
Hepatobiliary
Biliary tract carcinoma
<1
Hepatoblastoma
<1
Central nervous system
Congenital hypertrophy of the retinal pigmented epithelium (CHRPE)
75
Tumors (especially medulloblastoma)
<1
Two of these, duodenal cancer and desmoid disease, are major sources of morbidity and mortality (Fig. 37.3).
Other features may be a useful clue in diagnosis. CHRPE are hyper- or hypopigmented spots seen on retinal examination. They have no effect on vision but act as markers of FAP in the 66 % of families that have a total of at least four CHRPEs in both eyes.
Attenuated Familial Adenomatous Polyposis
A group of patients have been described who develop fewer than 100 colorectal adenomas (oligopolyposis) at a greater age (34–44 years) than in “classical” FAP, but who are at high risk of colorectal cancer, may exhibit extracolonic manifestations, and carry a germ line APC mutation.
The colorectal cancers have a later age of onset than with classical or profuse FAP (mean age 56 years).
The polyps have a rather different distribution, being more frequently found proximal to the splenic flexure, and their number varies significantly between family members, some of whom may have hundreds of adenomas.
The genotype of this group of patients may be one of the three: germ line APC mutation, biallelic MYH mutations, and germ line DNA mismatch repair (MMR) gene mutations.
APC mutations associated with attenuated familial adenomatous polyposis (AFAP) are either at in exons 3 and 4; at the 5′ end of the gene; or at the 3′ end of exon 15.
Fundic gland polyps (FGPs) and duodenal adenomas are frequent, but CHRPEs are not found in there patients.
Desmoid disease is rare in those with a 5′ mutation, but families with 3′ mutations (beyond about codon 1444) have a high risk of desmoid disease together with attenuated polyposis.
The missense APC mutation I1307K has been identified in Ashkenazi Jews with multiple adenomas, and E1317Q has also been found in association with AFAP.
When APC is normal, up to 30 % of patients with oligopolyposis have biallelic MYH mutations.
It is can be difficult to recognize AFAP clinically, leading to the clinical situation of an obstructing transverse colon cancer where right-sided polyposis is only found when the specimen is opened.
Because the polyps in AFAP are predominantly right sided, screening and work-up must include a full colonoscopy.
Genetic testing for germ line APC and MYH mutations has a relatively low yield, partially because of technical difficulties in detection of abnormalities that may be present and partly because gene expression may be lost for reasons other than a mutation.
A careful search (including upper gastrointestinal endoscopy) for extracolonic features of FAP, dye-spray colonoscopy to confirm polyp number, and testing of tumor or polyp tissue for microsatellite instability (MSI) and MMR immunohistochemistry (IHC) (to exclude Lynch syndrome) may be helpful.
Genetic testing for a germ line APC/MYH mutation should be pursued in patients with a total of ten or more colorectal adenomas, especially if there is a positive family history for colorectal adenomas or cancers.
A positive result has implication for family screening, but the patient is managed in the same way regardless of the result.
If the polyps are controllable endoscopically, then yearly colonoscopy is reasonable. If the adenoma burden is uncontrollable or dangerous, colectomy with ileorectal anastomosis (IRA) should be performed.
Gardner’s Syndrome
Gardner described the association between FAP and epidermoid cysts, osteomas, and “fibromas” (later found to be desmoid tumors) in 1953.
The term “Gardner’s syndrome” was later used to describe colorectal adenomatous polyposis occurring with these extracolonic manifestations.
Gardner’s syndrome is genetically the same as FAP, and systematic examination has revealed that most patients with FAP have at least one extraintestinal feature.
Though it is of historical interest, the term “Gardner’s syndrome” is no longer considered genetic or clinically useful and should be regarded as obsolete.
Turcot’s Syndrome
This is the association between colorectal adenomatous polyposis and central nervous system tumors. Recent molecular genetic investigation has shown that about two thirds of families have mutations in APC, with cerebellar medulloblastoma as the predominant brain tumor. Most of the other third, including Turcot’s original family, appear to be variants of hereditary nonpolyposis colorectal cancer (HNPCC) with glioblastoma as the predominant brain tumor and multiple (but fewer than 100) colorectal adenomas.
Presentation
Patients with FAP present either with or without symptoms (on screening).
There is a significant difference in cancer incidence between these two groups, with over 60 % of unscreened, symptomatic patients having colorectal cancer at presentation.
Screening
Clinical FAP screening begins at puberty because the risk of colorectal cancer under the age of 12 years is very small.
Genetic testing of at-risk family members in a family with a known mutation usually starts when endoscopic surveillance would start, at ages 12–14.
When a relative is identified as a mutation carrier, full colonoscopy is performed. EGD screening usually begins at age 20 years. Thyroid screening with ultrasound should also start then.
If genetic testing is uninformative or cannot be done in a family with classical FAP, endoscopic screening starts at age 12–14 with flexible sigmoidoscopy. Polyps are biopsied to prove they are adenomas.
An alternative would be to do retinal examinations for CHRPE or look for other extracolonic examinations with a skull X-ray or panorex examination of the jaw.
If a marker of FAP is found, full colonoscopy follows. The polyp burden is documented endoscopically and histologically and a decision made regarding the timing and type of surgery.
Symptoms
About 22 % of FAP patients have no family history. Clinical symptoms are often related to colorectal cancers, and should be investigated immediately.
Diagnosis
Genetic Testing
Genetic testing should be preceded by counseling, ideally by a genetic counselor. Counseling includes the provision of written information about the process and its consequences, after which informed consent is documented. The implications of genetic testing with respect to confidentiality, employment, insurance, and other financial issues vary from country to country but must be discussed prior to testing. In the USA, the Genetic Information Nondiscrimination Act (GINA) that became law in 2008 offers protection against genetic discrimination in Health and Life insurance. Posttest counseling deals with the implications of the genetic test results and may include psychological help to deal with emotional reactions, such as guilt (in an unaffected person), anxiety (in an affected person), and the effect of the results on family relationships.
DNA from an individual with clinically obvious FAP is sequenced to identify a mutation in APC, a process which is successful in about 80 % of cases. Failure to detect an APC mutation does not exclude a diagnosis of FAP and may occur for a variety of reasons, including the presence of large deletions or missense mutations. Such results have been misinterpreted as ruling out the diagnosis of FAP, with potentially serious consequences.
If a deleterious mutation is found in an affected family member, at-risk family members can be offered predictive testing with a high degree of accuracy. This is generally done between the ages of 12 and 15 years, when the individual is old enough to take part in genetic counseling.
When an individual does not carry the family mutation, that person can be discharged from further surveillance and be reassured that they do not have FAP.
A positive test result allows surveillance and prophylaxis to be targeted to those who need it, and knowledge of the site of mutation can aid decision making with regard to prophylactic surgery.
If no mutation can be found in an affected patient, then the family must be managed without genetic testing.
The negative result does not mean that the family does not have FAP; it means that the genetic cause of the FAP has not been found.
Management of the Large Bowel
Aims of Treatment. While the prevention of cancer remains an important priority in the management of patients with hereditary colorectal cancer, maintaining the quality of life is also important. This is especially the case in young, asymptomatic patients who have been diagnosed by screening. Where options exist for the timing and type of surgery, those with the least impact on social, academic, and vocational activities should be chosen. After all, surgery will not cure FAP.
Prophylactic Surgery. Patients with FAP, if untreated, are almost guaranteed to develop colorectal cancer.
Prevention of cancer by endoscopic control of the polyposis is not usually possible, and so colectomy or proctocolectomy is necessary to prevent cancer.
Timing. Patients with severe polyposis (over 1,000 colonic or over 20 rectal polyps), or those who are symptomatic, should have surgery as soon as possible.
In asymptomatic patients with mild disease (100–1,000 adenomas, all <1 cm, none with severe dysplasia), surgery can usually be delayed until the patient reaches appropriate physical and intellectual maturity.
An important reason for delay is the concern for the development of desmoid disease. Affected women with a family history of desmoid disease, extracolonic manifestations of Gardner’s syndrome, and a 3′ APC mutation are at highest risk.
As long as surgery is delayed, annual colonoscopy is recommended to monitor the polyps. Most patients with classical polyposis have surgery between the ages of 16 and 20, which is well before cancer usually develops.
Choice of Operation. The colorectal surgical options for the management of FAP are proctocolectomy with end ileostomy (with or without Koch pouch), colectomy with IRA, and proctocolectomy with ileoanal pouch (IPAA). Few patients desire a permanent ileostomy, and so proctocolectomy with ileostomy is rarely done.
IRA is more straightforward to perform than IPAA and requires only one procedure, with a shorter hospital stay and fewer complications. The risks of erectile and ejaculatory dysfunction caused by nerve damage during pelvic dissection are minimized, as is the significant reduction in fecundity observed in women after IPAA. In addition, bowel frequency and soiling are less, and no temporary stoma is necessary.
Polyp counts are a reliable way to identify a low-risk rectum, but patients still need yearly surveillance proctoscopy.
Any polyps over 5 mm should be removed, and polyps with high-grade dysplasia are relative indications for completion proctectomy.
Compared to an IRA, IPAA has the advantage of removing the entire colon and rectum. Although complication rates and functional results have improved with experience, they are still worse than those associated with IRA.
There has been controversy over the need for mucosectomy to remove the anorectal transition zone, which theoretically prevents cuff neoplasia, but causes more complications and perhaps poorer function. Dysplasia in the transition zone occurs after both double-stapled and mucosectomy techniques and the latter is probably only indicated in individuals with severe low rectal polyposis. The indications and contraindications and advantages and disadvantages of each surgical option are summarized in Table 37.3.
Table 37.3
Surgical options for familial adenomatous polyposis
Surgical option
Indication
Advantages
Disadvantages
Colectomy and ileorectal anastomosis (leave 15 cm rectum)
<20 rectal adenomas <1000 colon adenomas
Low complication rate
Risk of rectal cancer
No stoma
Close to normal bowel function
Proctocolectomy and ileal pouch anal anastomosis (stapled)
>20 rectal adenomas >1000 colon adenomas
Minimizes risk of rectal cancer
Complex surgery
Large rectal adenoma
Avoids permanent stoma
Often needs stoma
Rectal adenoma with severe dysplasia
Bowel function better than with mucosectomy and hand-sewn anastomosis
Bowel function unpredictable but may be quite abnormal
Sparing of low rectum
Risk of damage to pelvic nerves and decreased the ability of women to conceive
Risk of pouch and anal transitional adenomas and cancer
Proctocolectomy and ileal pouch anal anastomosis (hand sewn)
As above but with adenomas to dentate line
Minimizes risk of rectal cancer
As above but bowel function is worse than with stapled anastomosis
Avoids permanent stoma
Proctocolectomy with end ileostomy
Low rectal cancer
Simple operation with lower complication rate and minimal chance of reoperation
Permanent stoma
Poor anal sphincters
In summary, IRA is reasonable and safe in mildly affected patients, particularly if there are fewer than five rectal polyps.
Most individuals presenting with severe polyposis or those known to carry a mutation in codon 1309 should be advised to undergo IPAA.
But there are other issues. Pouch surgery in young men has an approximately 1 % risk of damage to erection, ejaculation, and bladder function; in women, fertility may be compromised.
Postoperative Surveillance
After IRA the retained rectum should be examined using a flexible sigmoidoscope, with a basic interval of 12 months or shorter, depending on the severity of disease. Polyps over 5 mm should be removed cleanly with a snare. Repeated polyp fulguration can result in rectal scarring, making future surveillance difficult and unreliable. In patients with chronically scarred rectal mucosa, random biopsy is recommended to detect invisible dysplasia. If severe dysplasia or uncontrolled polyposis develops, completion proctectomy with or without ileoanal pouch formation is indicated.
Surveillance of ileoanal pouches at several centers has shown adenomas in up to 53 % and even some cancers. Treatment of pouch adenomas depends on their number and size. Polyps over 5 mm should be removed by snare excision, while multiple small polyps respond to sulindac (150 mg by mouth twice daily).
Anal transition zone (ATZ) adenomas occur commonly after both stapled and hand-sewn IPAA, although they are twice as common in the former as the latter.
Several case reports of cancer in the ATZ underline the difficulty in following this critical area.
Adenomas in the ATZ can be excised individually (under anesthesia), or the entire ATZ can be stripped. If stripping is chosen because of the extent of the polyposis, the procedure should be performed in two stages to avoid stenosis.
Adenoma Chemoprevention
A range of chemopreventive agents have been studied in FAP, in part because of the problems of managing the retained rectum after IRA, but also because this disease provides a useful experimental model of colorectal carcinogenesis. In placebo-controlled trials, both the nonsteroidal anti-inflammatory drugs (NSAIDs) sulindac and the COX-2 inhibitor celecoxib have reduced the number and size of colorectal adenomas.Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree






